00017793722021FYFALSE0.3636P5D00017793722021-01-012021-12-310001779372us-gaap:CommonClassAMember2021-01-012021-12-310001779372us-gaap:WarrantMember2021-01-012021-12-3100017793722021-11-11iso4217:USD00017793722022-03-23xbrli:shares00017793722021-12-3100017793722020-12-31iso4217:USDxbrli:shares00017793722020-01-012020-12-310001779372us-gaap:CommonStockMember2019-12-310001779372us-gaap:AdditionalPaidInCapitalMember2019-12-310001779372us-gaap:RetainedEarningsMember2019-12-3100017793722019-12-310001779372us-gaap:AdditionalPaidInCapitalMember2020-01-012020-12-310001779372us-gaap:CommonStockMember2020-01-012020-12-310001779372us-gaap:RetainedEarningsMember2020-01-012020-12-310001779372us-gaap:CommonStockMember2020-12-310001779372us-gaap:AdditionalPaidInCapitalMember2020-12-310001779372us-gaap:RetainedEarningsMember2020-12-310001779372us-gaap:AdditionalPaidInCapitalMember2021-01-012021-12-310001779372us-gaap:CommonStockMember2021-01-012021-12-310001779372us-gaap:RetainedEarningsMember2021-01-012021-12-310001779372us-gaap:CommonStockMember2021-12-310001779372us-gaap:AdditionalPaidInCapitalMember2021-12-310001779372us-gaap:RetainedEarningsMember2021-12-31beat:segment00017793722021-09-2700017793722021-09-272021-09-27xbrli:pure00017793722021-11-012021-11-300001779372us-gaap:SubsequentEventMember2022-02-012022-02-280001779372us-gaap:StockOptionMember2021-01-012021-12-310001779372us-gaap:StockOptionMember2020-01-012020-12-310001779372us-gaap:WarrantMember2021-01-012021-12-310001779372us-gaap:WarrantMember2020-01-012020-12-310001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMember2015-08-210001779372beat:QualifiedFinancingMemberbeat:AmendmentNumberSixMemberus-gaap:ConvertibleDebtMember2021-03-220001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMemberbeat:AmendmentNumberSevenMember2021-10-070001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMember2021-12-310001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMember2021-11-102021-11-100001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMember2021-11-100001779372beat:QualifiedFinancingMemberus-gaap:ConvertibleDebtMember2021-09-300001779372beat:QualifiedFinancingMemberus-gaap:ConvertibleDebtMember2021-07-012021-09-300001779372beat:QualifiedFinancingMemberus-gaap:ConvertibleDebtMember2021-12-310001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMember2020-12-3100017793722020-10-012020-12-310001779372us-gaap:ConvertibleDebtMemberbeat:ShortTermNotesPayableMember2020-01-012020-12-310001779372beat:A2015NotesMemberus-gaap:ConvertibleDebtMember2020-01-012020-12-310001779372us-gaap:ConvertibleDebtMember2020-01-012020-12-310001779372beat:PromissoryNoteMemberus-gaap:ConvertibleDebtMember2020-01-012020-12-310001779372us-gaap:ConvertibleDebtMemberbeat:AdvanceMember2020-01-012020-12-310001779372us-gaap:ConvertibleDebtMemberbeat:AccruedInterestMember2020-01-012020-12-310001779372beat:A3PromissoryNoteMember2020-12-3100017793722020-04-212020-04-210001779372us-gaap:IPOMember2021-11-102021-11-100001779372us-gaap:IPOMemberus-gaap:CommonStockMember2021-11-102021-11-100001779372us-gaap:IPOMemberus-gaap:CommonStockMember2021-11-100001779372us-gaap:IPOMemberus-gaap:WarrantMember2021-11-102021-11-100001779372us-gaap:IPOMemberus-gaap:WarrantMember2021-11-100001779372beat:ConsultantsMemberus-gaap:IPOMemberus-gaap:CommonStockMember2021-11-102021-11-100001779372beat:ConsultantsMemberus-gaap:IPOMemberus-gaap:WarrantMember2021-11-102021-11-100001779372beat:ConsultantsMemberus-gaap:IPOMemberus-gaap:CommonStockMember2021-11-1000017793722019-01-012019-12-310001779372beat:PennyWarrantMember2019-12-310001779372beat:PennyWarrantMember2019-01-012019-12-310001779372us-gaap:IPOMember2021-11-100001779372us-gaap:OverAllotmentOptionMember2021-11-102021-11-100001779372us-gaap:OverAllotmentOptionMember2021-11-1000017793722021-11-1500017793722021-11-152021-11-1500017793722021-01-012021-03-310001779372beat:A2015EquityIncentivePlanMember2015-12-310001779372beat:A2015EquityIncentivePlanMember2018-01-312018-01-310001779372beat:A2015EquityIncentivePlanMember2021-06-152021-06-150001779372beat:A2015EquityIncentivePlanMember2021-12-310001779372srt:MinimumMemberus-gaap:EmployeeStockOptionMember2021-01-012021-12-310001779372us-gaap:EmployeeStockOptionMembersrt:MaximumMember2021-01-012021-12-310001779372srt:MinimumMemberus-gaap:EmployeeStockOptionMember2020-01-012020-12-310001779372us-gaap:EmployeeStockOptionMembersrt:MaximumMember2020-01-012020-12-310001779372us-gaap:EmployeeStockOptionMember2021-01-012021-12-310001779372us-gaap:EmployeeStockOptionMember2020-01-012020-12-310001779372us-gaap:GeneralAndAdministrativeExpenseMember2021-01-012021-12-310001779372us-gaap:GeneralAndAdministrativeExpenseMember2020-01-012020-12-310001779372us-gaap:ResearchAndDevelopmentExpenseMember2021-01-012021-12-310001779372us-gaap:ResearchAndDevelopmentExpenseMember2020-01-012020-12-310001779372us-gaap:RestrictedStockUnitsRSUMember2021-12-142021-12-140001779372us-gaap:RestrictedStockUnitsRSUMember2021-12-310001779372beat:AccountingServicesMembersrt:AffiliatedEntityMemberbeat:CTRLCFOAndHardestyMember2021-01-012021-12-310001779372beat:AccountingServicesMembersrt:AffiliatedEntityMemberbeat:CTRLCFOAndHardestyMember2020-01-012020-12-310001779372beat:AccountingServicesMembersrt:AffiliatedEntityMemberbeat:CTRLCFOAndHardestyMember2021-12-310001779372beat:AccountingServicesMembersrt:AffiliatedEntityMemberbeat:CTRLCFOAndHardestyMember2020-12-310001779372beat:A2015NotesInvestmentsMembersrt:ManagementMemberbeat:DirectorsAndOfficersMember2021-11-102021-11-100001779372beat:A2015NotesMemberbeat:A2015NotesInvestmentsMembersrt:ManagementMemberbeat:DirectorsAndOfficersMember2021-11-102021-11-100001779372beat:ConsultantsMemberbeat:A2015NotesInvestmentsMember2021-11-102021-11-100001779372beat:A2015NotesInvestmentsMembersrt:ManagementMemberbeat:DirectorsAndOfficersMember2020-01-012020-12-310001779372beat:ConsultantsMemberbeat:A2015NotesInvestmentsMember2020-01-012020-12-310001779372us-gaap:DomesticCountryMember2021-12-310001779372us-gaap:StateAndLocalJurisdictionMember2021-12-310001779372us-gaap:PrivatePlacementMemberbeat:OpenSkyOpportunitiesFundLtdMemberus-gaap:SubsequentEventMember2022-02-182022-02-180001779372us-gaap:PrivatePlacementMemberus-gaap:CommonStockMemberbeat:OpenSkyOpportunitiesFundLtdMemberus-gaap:SubsequentEventMember2022-02-182022-02-180001779372us-gaap:PrivatePlacementMemberus-gaap:CommonStockMemberbeat:OpenSkyOpportunitiesFundLtdMemberus-gaap:SubsequentEventMember2022-02-180001779372us-gaap:WarrantMemberus-gaap:PrivatePlacementMemberbeat:OpenSkyOpportunitiesFundLtdMemberus-gaap:SubsequentEventMember2022-02-182022-02-180001779372us-gaap:PrivatePlacementMemberbeat:OpenSkyOpportunitiesFundLtdMemberus-gaap:SubsequentEventMember2022-02-180001779372us-gaap:WarrantMemberus-gaap:PrivatePlacementMemberbeat:OpenSkyOpportunitiesFundLtdMemberus-gaap:SubsequentEventMember2022-02-180001779372us-gaap:SubsequentEventMember2022-03-070001779372us-gaap:SubsequentEventMemberbeat:LIVMORAndTripleRingMember2022-01-312022-03-07
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| | | | | |
| ☒ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2021
or
| | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from ___________ to ___________
Commission File Number: 001-41060
HEARTBEAM, INC.
(Exact Name of Registrant as Specified in its Charter)
| | | | | | | | |
| Delaware | | 47-4881450 |
State or Other Jurisdiction of Incorporation or Organization | | I.R.S. Employer
Identification No. |
| | |
2118 Walsh Avenue, Suite 210 Santa Clara, CA 95050 | | 95050 |
| Address of Principal Executive Offices | | Zip Code |
(408) 899-4443
Registrant’s Telephone Number, Including Area Code
Securities registered pursuant to Section 12(b) of the Act:
| | | | | | | | | | | | | | |
| Title of each class | | Trading Symbol(s) | | Name of each exchange on which registered |
| Common Stock | | BEAT | | NASDAQ |
| Warrants | | BEATW | | NASDAQ |
Securities registered pursuant to section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☒
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | |
Large accelerated filer ☐ | Accelerated filer ☐ |
Non-accelerated filer ☒ | Smaller reporting company ☒ |
| Emerging growth company ☒ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☒
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant, based on the closing price of a share of the registrant’s common stock as reported by the NASDAQ Capital Market on such date was approximately $26,617,634. The registrant has elected to use November 11, 2021 which was the initial trading date on the NASDAQ Capital Market, as the calculation date because on June 30, 2021 (the last business day of the registrant’s mostly recently completed second fiscal quarter), the registrant was a privately-held company. Shares of the registrant’s common stock held by each executive officer, director and holder of 5% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates. This calculation does not reflect a determination that certain persons are affiliates of the registrant for any other purpose.
As of March 23, 2022, there was 7,958,888 shares of the registrant’s common stock issued and outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of HeartBeam, Inc.’s Proxy Statement in connection with its Annual Meeting of Stockholders to be held on June 15, 2022 are incorporated by reference into Part III of this report.
HEARTBEAM, INC.
CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains “forward-looking statements” within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). In particular, statements contained in this Annual Report on Form 10-K, including but not limited to, statements regarding the sufficiency of our cash, our ability to finance our operations and business initiatives and obtain funding for such activities; our future results of operations and financial position, business strategy and plan prospects, or costs and objectives of management for future acquisitions, are forward looking statements. These forward looking statements relate to our future plans, objectives, expectations and intentions and may be identified by words such as “may,” “will,” “should,” “expects,” “plans,” “anticipates,” “intends,” “targets,” “projects,” “contemplates,” “believes,” “seeks,” “goals,” “estimates,” “predicts,” “potential” and “continue” or similar words. Readers are cautioned that these forward-looking statements are based on our current beliefs, expectations and assumptions and are subject to risks, uncertainties, and assumptions that are difficult to predict, including those identified below, under Part II, Item lA. “Risk Factors” and elsewhere in this. Therefore, actual results may differ materially and adversely from those expressed, projected or implied in any forward-looking statements. We undertake no obligation to revise or update any forward looking statements for any reason.
The Company will continue to file annual, quarterly and current reports, proxy statements and other information with the Securities and Exchange Commission (the “SEC”). Forward-looking statements speak only as of the dates specified in such filings. Except as expressly required under federal securities laws and the rules and regulations of the SEC, we do not undertake any obligation to update any forward-looking statements to reflect events or circumstances arising after any such date, whether as a result of new information or future events or otherwise. You should not place undue reliance on the forward-looking statements included in this report or that may be made elsewhere from time to time by us, or on our behalf. All forward-looking statements attributable to us are expressly qualified by these cautionary statements.
NOTE REGARDING COMPANY REFERENCES
Throughout this Annual Report on Form 10-K, “HeartBeam,” the “Company,” “we,” “us” and “our” refer to HeartBeam, Inc.
Table of Contents
| | | | | |
| Page |
| CAUTIONARY STATEMENT REGARDING FORWARD-LOOKING STATEMENTS | |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
| |
Part IV | |
| |
| |
| |
Part I
Item 1. Business
Overview
We are a medical technology company primarily focusing on telemedicine solutions that enable the detection and monitoring of cardiac disease outside a healthcare facility setting. Our aim is to deliver innovative, remote diagnostic and monitoring technologies that can be used for patients anywhere, with initial offerings for ambulatory and emergency department use. Our products require Food and Drug Administration (“FDA”) clearance and have not been cleared for marketing (hereinafter “Product” or “Products”.) We believe our Products and services will benefit many stakeholders, including patients, healthcare providers, and healthcare payors. Our initial focus is providing diagnostic data to help physicians with care management of patients with cardiovascular disease. There are two major markets for our initial Products: remote patient monitoring and the hospital Emergency Department (“ED”). We are developing our telehealth Product to address the rapidly growing field of remote patient monitoring. Our telehealth Product is comprised of a credit card sized Electrocardiogram (“ECG”) machine and a powerful cloud-based diagnostic software expert system. We believe that we are uniquely positioned to play a central role in remote monitoring of high-risk coronary artery disease patients, because the studies performed so far have shown that our ischemia detection system is highly accurate. Our powerful ischemia detection system is unlike other ambulatory cardiac monitors currently on the market which focus on arrhythmia detection. Also, we are applying our platform technology to create a software tool for detecting heart attacks in the ED environment. This software tool is designed to enable emergency physicians to more accurately and quickly diagnose heart attacks than currently available. Market release of this Product will precede that of the telehealth Product.
To date, we have developed working prototypes for both our telehealth Product and our ED Product. The ED Product is currently undergoing additional engineering work that we believe will make it ready for FDA 510(k) clearance submission. Both Products have been validated in three medical studies, which were designed by Harvard Medical School faculty. Peer reviewed publications that describe the studies and results are in preparation. One peer reviewed medical publication submission is planned for the second quarter of 2022 and followed by another one later in 2022. These two publications will describe results of our two key studies: HeartBeam Ischemia Detection Study (“HIDES”) and B Score. In 2022 we plan to publish results of a study on performance of our ED Product in the real-life environment of an ED. In November 2021 we presented a technology abstract at the IEEE EMBS 2021 Conference. The abstract describes the technology foundation of our ED Product.
The custom software and hardware of our Products, we believe, are classified as Class II medical devices by the FDA running on an FDA approved Class I registered platform. Class II medical devices are those for which general controls alone are insufficient to provide reasonable assurance of safety and effectiveness and there is sufficient information to establish special controls. Special controls can include performance standards, post-market surveillance, patient histories and FDA guidance documents. Premarket review and clearance by the FDA for these devices is generally accomplished through the 510(k) or 510(k) de-novo premarket notification process.
HeartBeam has three issued U.S. patents (U.S.10,433,744, U.S.10,117,592 and U.S.11,071,490), and six pending U.S applications. Three of the pending applications have published, all of the remaining three pending cases are unpublished. Outside of the U.S., HeartBeam has three pending European (“EU”) utility patent applications (one of which is allowed), one pending Canadian (“CA”) utility patent application, one pending Chinese (“CN”) utility patent application, one pending Japanese (“JP”) utility patent application, one pending Australian (“AU”) application, and two pending Patent Cooperation Treaty (“PCT”) applications.
Market Overview
Chronic diseases are the number one burden on the healthcare system, driving up costs each year, and cardiovascular illnesses are one of the top contributors. Regulators, payors and providers are focused on shifting the diagnosis and management of these conditions to drive better outcomes at lower cost. Connected medical solutions are expanding rapidly and are projected to reach $155 billion by 2026, a compound annual growth rate (“CAGR”) of 17%. These solutions are socio-technical models for healthcare management and delivery using technology to provide healthcare services remotely and aim to maximize healthcare resources and provide increased, flexible opportunities for consumers to engage with clinicians and better self-manage their care, using readily available consumer technologies to deliver patient care outside of the hospital or doctor’s office. The types of companies that make up this market include Accenture, IBM, SAP, GE
Healthcare, Oracle, Microsoft, Airstrip Technology, Medtronic, Allscripts, Boston Scientific, Athenahealth, Cerner, Philips, Agamatrix, Qualcomm, and AliveCor.
The market for remote patient monitoring (“RPM”), is projected to reach $31.3 billion by 2023. In 2019, 1,800 hospitals in the US were using mobile applications to improve risk management and quality of care. We believe the number in 2020 was likely larger as the onset of the COVID-19 pandemic greatly accelerated use and acceptance of telehealth by both patients and healthcare providers.
Cardiovascular disease is the number one cost to the healthcare system and is estimated to be responsible for 1 in every 6 healthcare dollars spent in the US. As cardiovascular disease is the leading cause of death worldwide, early detection, diagnosis, and management of chronic cardiac conditions are necessary to relieve the increasing burden on the healthcare infrastructure. Diagnostic tests such as ECGs are used to detect, diagnose and track numerous cardiovascular conditions. With advances in mobile communications, diagnostic monitoring of cardiac conditions is increasingly occurring outside the hospital. We believe that the trend of moving diagnostic data collection and care for heart disease to the ambulatory setting is a permanent change. This trend will continue to place increased value on cost-effective diagnostic and monitoring technologies in the ECG market.
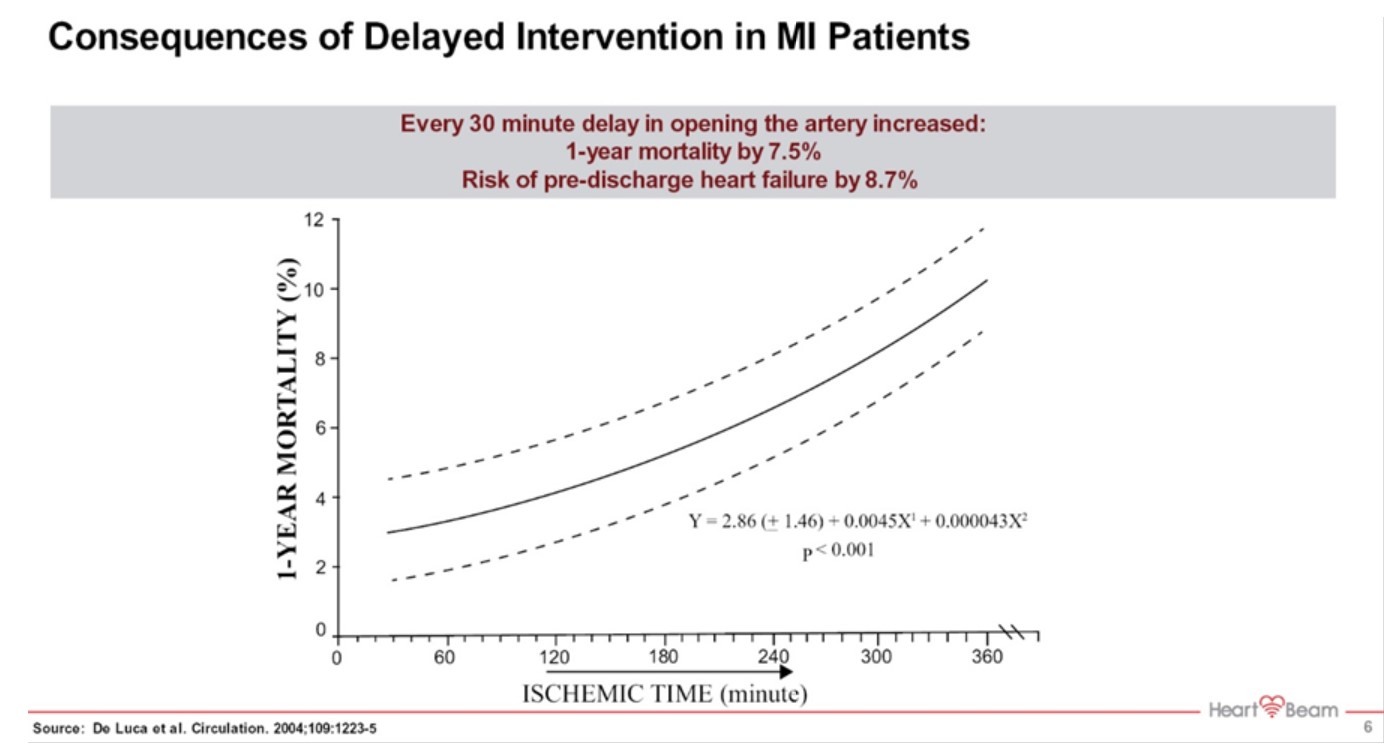
Time to intervention is one the key factors that influence medical outcome for a patient that is experiencing a heart attack. Yet, for a number of reasons, on average, a heart attack patient will wait about three hours before seeking help. That has very negative consequences on mortality rate for these patients as well as a marked increase in the heart failure incidence in these patients. The huge increase in healthcare expenditures associated with that delay in seeking medical help is responsible for relative increase in heart failure rate of 8.7% for every 30 minutes of delay. Heart failure is one of the most expensive diagnoses to treat.
Our initial telemedicine technology Product will address the heart attack detection market as well as the market to monitor Coronary Artery Disease (“CAD”) patients who are typically at high risk for a heart attack. Currently there are no products on the market that are user friendly, easy to carry, and always with the patient in order to provide physicians and patients with timely and highly accurate information about potential Acute Coronary Syndrome (“ACS”) and Myocardial Infarction (“MI”) events. A tool that is always with the patient, that decreases time to intervention, and that decreases the number of unnecessary ED visits by chest pain patients would have a significant effect on saving lives and healthcare dollars. We believe our technology will address this problem and will provide a convenient, cost-effective, integrated telehealth solution, including software and hardware for physicians and their patients. There are approximately 18 million people in
the US who are considered at high risk for a heart attack, including 8 million who already have had prior intervention for MI’s and are therefore considered to be at extreme risk.
In the US, mobile cardiac tests are primarily conducted through outsourced Independent Diagnostic Testing Facilities (“IDTFs”) or as part of a RPM system. Reimbursement rates vary depending on the use case and generally are based on the value a technology offers to patients and healthcare providers. Actual reimbursed pricing is set by the Centers for Medicare & Medicaid Services (“CMS”). Reimbursement rates for private insurers typically provide for similar or better reimbursement rates when compared to those set by the Government for Medicare and Medicaid.
In the ED environment, early and accurate diagnosis of a chest pain patient who is potentially having a heart attack is of immense importance. Guidelines state that every chest pain patient in an ED must receive an ECG within 10 minutes of presentation. The interpretation accuracy of these initial ECGs is only approximately 66%. The need for increased ECG accuracy in detecting a heart attack in the ED is well defined, and an improved solution could result in saved lives and healthcare dollars. We are currently developing a version of our ED Product that will be hosted by LIVMOR, Inc., a digital health solutions company (“LIVMOR”) on a white label version of its FDA Class 1 registered platform. Our ED Product is being developed to meet all requirements for FDA 510(k) clearance submission and we plan to submit this Product for FDA clearance during the second quarter of 2022. We believe this Product will offer a marked increase in the accuracy of heart attack detection in EDs. There are approximately 5,000 ED departments in the US and 137 million ED visits per year.
Products and Technology
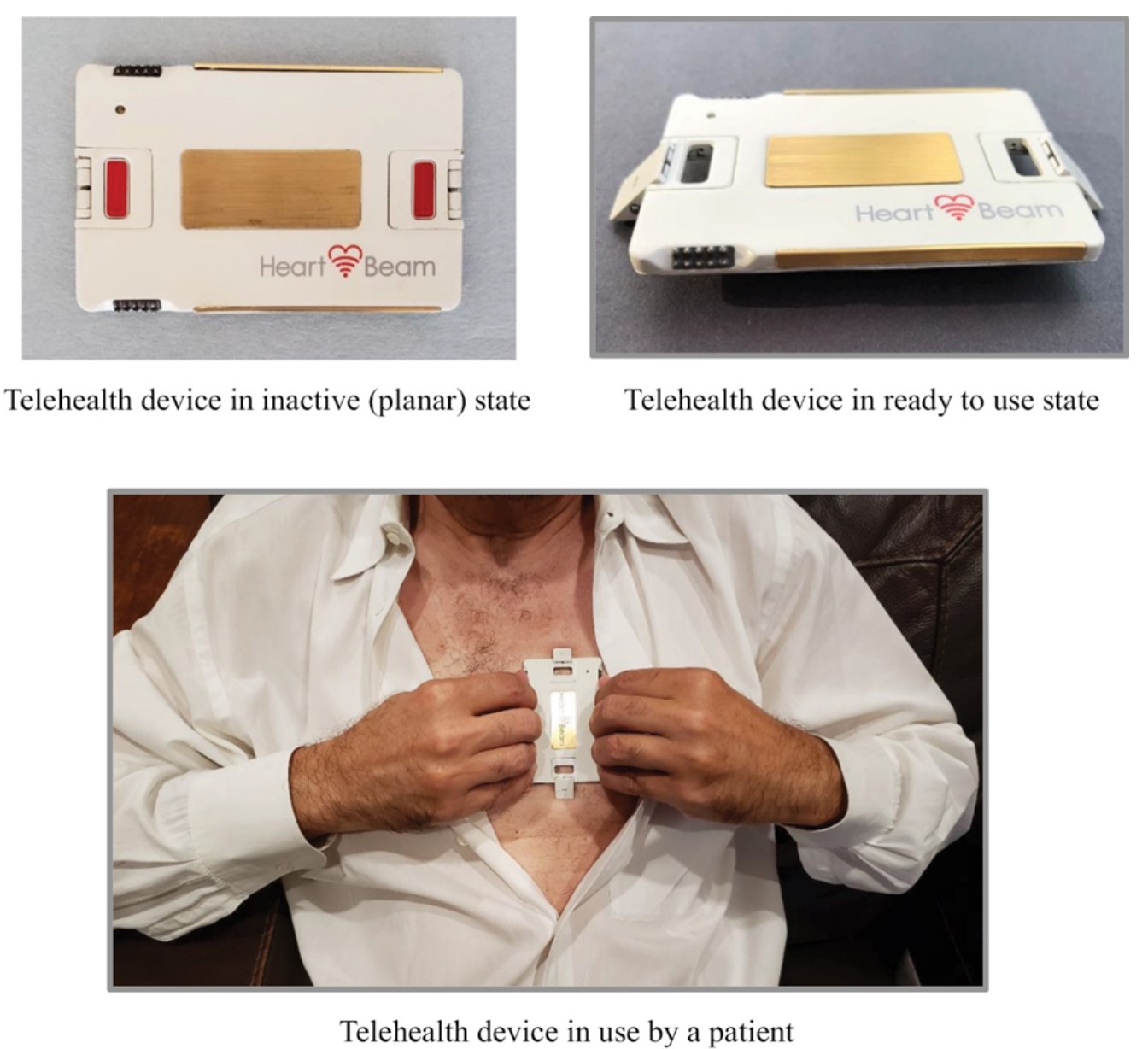
The foundation of our novel technology is the concept of vectorcardiography (“VCG”), a technology that has long been seen as superior to ECGs in detecting MIs, but is no longer used clinically because of the difficulty experienced by physicians interpreting the output. We solved the crucial problem of recording three orthogonal (x, y and z) projections of the heart vector with a device that is sized like a credit card. The thickness of our credit card sized ECG signal collection device is about 1/8 inch (3 mm) and it weighs about 1 ounce (28 grams).
The core technology consists of a series of patented inventions and associated algorithms. In addition to using VCG to get a more complete 3D characterization of cardiac activity, we use the concept of a baseline. Our MI detection marker is a differential marker that measures the change in cardiac parameters between an asymptomatic (baseline) recording and the symptomatic recording. It is personalized for every patient as every patient has a unique baseline. Our increase in diagnostic performance in detecting MIs, when compared to a panel of cardiologists, is attributed to a richer cardiac information set offered by VCG and the fact that our MI marker compares the baseline and symptomatic recordings and does that in the 3D space of VCG.
This novel technology has resulted in two key Products to date: a telehealth Product for high-risk cardiovascular patients and a powerful cloud-based diagnostic expert and MI detection system for EDs. Our telehealth ECG collection device is the size of a credit card and records cardiac signals with integrated electrodes rather than wires or self-adhesive electrodes. Unlike a standard 12-lead ECG machine that records signals in empirically determined locations on a human body, our approach is focused on recording three projections of the heart vector. The successful recording of the projections of the heart vector enables the synthesis of a 12-lead signal set as well as internal algorithmic diagnostic work in the space of 3D heart vectors.
There are obvious ease of use advantages when comparing our handheld device that fits in a wallet and can be instantly self-applied versus the current 12-lead ECG machine that requires a trained professional to apply. In addition, there are diagnostic performance advantages, including, based on our initial study, increased accuracy in diagnosing MIs. The system is used by patients at home or elsewhere, with help from their physicians, to assess whether their chest pain is truly the result of an MI.
Our telehealth system will be a prescription-only mobile health system intended for individuals with known or suspected heart disease, especially CAD. It helps guide physicians in choosing the best course of action for their patients who experience chest pain outside of a medical facility. Our system will bring a medical grade ECG to patients and will enable them to receive a plan of action from a physician in a timely manner. At the time of onboarding and at regularly scheduled time intervals, patients record a baseline 30 second cardiac reading using our device. When a patient experiences symptoms, such as irregular heartbeats or chest pain, the patient can simply open the smartphone app and press the credit card sized device against the chest to collect signals that can be converted to a synthesized 12-lead ECG. This derived 12-lead ECG is sent to the physician overlayed with the patient’s derived baseline ECG recording. In addition, the patient provides input on their symptoms that are sent, along with the ECG data, to the cloud for interpretation. A cloud-based algorithm processes the signals and displays the symptom description and patient history to a physician to analyze and prescribe an action plan. From start to finish, the process takes just a few minutes.
The telehealth system consists of:
1.A credit card sized cardiac electrical signal collection device. The device captures cardiac signals that represent x, y and z projections of the heart vector and transmits them via Bluetooth connection to a smartphone. It is always with the patient as it easily fits in a wallet. It is easy to use as all that is required of the patient is that the device be pressed against the chest.
2.A smartphone application that receives the cardiac signals from the HeartBeam signal collection device. The app has several functions: guiding the patient through the signal collection, asking about symptoms, displaying the status of the data collection, and notifying the patient of the plan of action as determined by a physician. In addition, the app will contain HIPAA-compliant video conferencing or text capabilities for the healthcare provider to communicate directly with the patient.
3.A cloud-based software system that serves three basic functions: (1) Performing a final check of the ECG signal quality, (2) Synthesizing a 12-lead ECG from the measured (recorded) 3 vector leads, and (3) Preparing a summary report for the physician. In order to facilitate a more accurate physician interpretation of the data, the software overlays the patient’s synthesized baseline 12 lead ECG waveform on the synthesized 12 lead ECG waveform from the current event. To ensure high signal quality, the system checks for noise levels in the recorded signals. Those signals that can be effectively filtered are accepted and those that have a noise level above an empirically established threshold are rejected. If a recorded signal is rejected, the user is asked to repeat the recording.
4.A web-based physician portal, which displays all of the relevant information for the physician to analyze: patient history, symptoms, baseline and current readings, synthesized 12 lead ECG, and recorded 3 vector leads. The HeartBeam physician portal assists physicians with their diagnostic interpretation by providing both the baseline 12-lead synthesized ECG and the 12-lead synthesized ECG that is under evaluation.
The market release of our telehealth Product will be in two generations. The Generation 1 product will have a limited feature set and will offer to the physician a pair of baseline and symptomatic ECGs and a symptoms report. This product is an excellent match for existing current procedural terminology (CPT) remote patient monitoring reimbursement codes. The Generation 2 product will feature our proprietary MI marker as well as our diagnostic suggestion in addition to all features of Generation 1 product. Since Generation 2 will offer increased medical value, we plan to seek a unique reimbursement code for this product.
The same core technology is used in the ED Product. In this application, the increased accuracy of detecting MIs by the ECG is of utmost importance. An ECG is the first diagnostic test a chest pain patient receives in the ED and it has a major impact on the patient’s subsequent clinical path. The ED Product has no hardware and introduces minimal change to the standard of care chest pain diagnostic path. It uses a baseline standard 12-lead ECG from the patient’s Electronic Medical Record (EMR) and the chest pain ECG that is being evaluated. It converts both of them to a VCG representation and utilizes our proprietary 3D VCG differential marker. An initial clinical study indicates that the ED software Product offers considerable improvement in the accuracy of MI detection compared to a panel of experienced cardiologists interpreting a standard 12 lead ECG. The importance of increased accuracy of the first ECG interpretation for a chest pain patient presenting to an ED is significant, potentially leading to faster intervention or avoiding unnecessary activation of the cardiac catheterization lab.
FDA Regulatory Path
We have defined the FDA 510(k) clearance paths for both Products and have contracted with regulatory consultants to help us clear both products with the FDA.
Two predicate devices were identified that combined, we believe, will be acceptable to the FDA for our telehealth Generation 1 Product submission. The submission is planned to contain results of a validation study. The nature of this study will be a comparison of our synthesized 12-lead ECG recordings to standard 12-lead ECG recordings and to the synthesized ECG recordings from one of the predicate devices. We plan to submit our Generation 1 telehealth product to the FDA for a 510(k) clearance in the fourth quarter of 2022.
The ED software product is hosted on a white label version of LIVMOR’s Class I registered software platform and a predicate device for the cloud-based diagnostic engine for the ED product was identified. It is widely used as it is part of a software package that one of the leading ECG machine manufacturers offers. The predicate device software makes a diagnostic suggestion in regard to a potential MI diagnosis. Our ED software Product will also make a diagnostic suggestion to the ED physician. In our HIDES study we have shown superiority in detecting ischemia (ACS) over a panel of cardiologists. The study that will be used for the FDA 510(k) clearance application will be a retrospective study of patient's electronic medical records that contain the patient's chest pain ECGs at the presentation to the ED and their baseline asymptomatic ECG recordings. The diagnostic suggestion of the predicate device software, which is already contained in the EMR, will be compared to the diagnostic suggestion of our Product. We plan to submit our ED Product for FDA 510(k) clearance in the second quarter of 2022.
Market Opportunity
ECGs are key diagnostic tests utilized in the diagnosis and monitoring of cardiovascular disease, the number one cause of death worldwide. In the US in 2016, there were 121.5 million adults living with cardiovascular disease and 18.3 million adults with diagnosed coronary artery disease. The market size is increasing, due to an aging population and lifestyle choices.
Every 40 seconds someone in the US has a heart attack, or MI. Unfortunately, there is no way for patients to tell whether the symptoms they are experiencing are due to an MI, or some other more benign condition such as indigestion. As a result, patients often ignore symptoms and delay seeking care, which leads to worse outcomes and increased mortality. On the other hand, many patients who go to the ED with chest pain are not experiencing an MI. Chest pain is the second most
common reason for an ED visit, yet fewer than 20% of chest pain ED visits result in a diagnosis of a life-threatening condition. These unnecessary ED visits lead to well over $10 billion in unnecessary healthcare expenditures.
Most ECGs are conducted in a healthcare facility setting using a 12-lead ECG machine, the gold standard. ECGs taken outside of healthcare facilities are expected to grow more quickly than in-hospital ECGs. Monitoring cardiac patients outside of a hospital is a fast growing trend, as it is less expensive and provides a better patient experience. However, while ambulatory cardiac monitoring devices are often much easier for patients to use, they have fewer leads than the gold standard and therefore cannot offer as comprehensive a picture of cardiac health.
While a standard 12-lead ECG readout is of great medical value, it is simply impractical to have a machine next to patients when they experience symptoms outside the clinical setting, since recording the event requires attaching multiple electrodes to the patient’s body with professional assistance. While existing technologies use predominantly single lead ECG devices to monitor arrhythmias, these technologies do not provide information to the physician on the presence of life threatening conditions of ACS or heart attacks.
We believe our telehealth technology addresses these market needs and has several key attributes that make it a good fit for these patients. Our telehealth Product is generally used when symptoms occur and offers the potential for lifelong patient usage. The device is always near the patient and ready to be used for recording a cardiac event. It enables real-time cardiac data transmission during a telemedicine visit. It offers a recorded 3 vector lead set of signals and a 12-lead derived ECG set of signals. Physicians will typically prescribe our solution to chronic cardiovascular patients for long term monitoring, thereby enabling prolonged data collection and delivering a more complete picture for diagnosis. This will also enable the use of artificial intelligence on our future database that will have a unique set of longitudinal 12-lead ECGs for patients.
Market Strategy
Our goal is to establish our products as key solutions for cardiology practices and hospital EDs. Our efforts to enter the market involve establishing clinical evidence and demonstrating the cost-effectiveness of adopting our products. For both the telehealth and the ED Products, the initial geographic market is the United States.
We believe that both the telehealth and EDs for our ED Product and Generation 1 telehealth Product. For the telehealth Product, the primary customers are cardiology practices and the cardiology departments of hospitals. Healthcare insurers are another important customer, as they will benefit from the reduced costs to the healthcare system. We are working to develop new clinical studies and publish results of completed clinical studies and plan to demonstrate real world cost-effectiveness of the use of the solution.
A key element of our strategy is obtaining reimbursement for the telehealth Product. This strategy has two stages in full alignment with our Generation 1 and Generation 2 telehealth product introduction plans. In the short term, we expect that physicians will use existing Remote Patient Monitoring (“RPM”) reimbursement codes. We believe that our Generation 1 telehealth Product will be a compelling offering among RPM technologies, as it will be uniquely positioned to assess ACS and heart attacks among high-risk cardiac patients. In the longer term, we will conduct additional clinical trials that demonstrate the clinical efficacy and cost effectiveness of our next generation, full featured Generation 2 Product and will work to secure a new CPT code and reimbursement specific to the Generation 2 telehealth solution. RPM codes pay practices for providing covered services.
The main RPM codes relevant for our Generation 1 telehealth Product are:
•CPT 99453: Remote monitoring of physiologic parameter(s), initial; set-up and patient education on use of equipment
•CPT 99454: Remote monitoring of physiologic parameter(s), initial; device(s) supply with daily recording(s) or programmed alert(s) transmission, each 30 days
•CPT 99457: Remote physiologic monitoring treatment management services, clinical staff/physician/ other qualified health care professional time in a calendar month requiring interactive communication with the patient/caregiver during the month; initial 20 minutes
•CPT 99458: Remote physiologic monitoring treatment management services, clinical staff/physician/ other qualified health care professional time in a calendar month requiring interactive communication with the patient/caregiver during the month; additional 20 minutes
CPT 99453 is paid one-time per patient, with the average CMS payment rate of $21. The technical code CPT 99454 and the professional code CPT 99457 are paid monthly, with a combined average CMS payment rate of $119. Private payers may pay at different amounts.
Practices will bill payers for monthly services related to the core HeartBeam telehealth Product, potentially bundled with a third-party blood pressure measurement device. Under this model, the company will negotiate with payers for a per patient per month fee for the ongoing HeartBeam telehealth service, which also will include an amortized charge for the cost of the device.
We will explore other business models. For example, hospitals face CMS penalties if their 30-day readmission rates for patients who are discharged after an MI exceed certain thresholds. These CMS penalties are levied on all hospital CMS payments, so the impact can be significant. Our telehealth Product can be a tool to help hospitals manage these patients after discharge. We will explore models in which hospitals pay for the device and for the initial 30 days of service. In addition, we will explore models for value-based care, in which the use of the telehealth Product reduces overall costs.
We have engaged two senior sales executives in Atlanta and Denver with extensive experience in medical device sales. We are currently speaking with hospitals in large healthcare systems to educate them about our first two products. These sales executives will be responsible for building a direct sales force for our telehealth Product and to target large hospitals and integrated practices. These are sophisticated customers, and we plan to use technical presentations, peer reviewed clinical data, and demonstration projects to achieve penetration of this market. We plan to continue to expand our medical advisory board, conduct clinical trials with leading cardiologists to increase the body of evidence, and establish reference sites among these customers.
Our long-term strategy is to generate sufficient evidence of clinical efficacy and cost-effectiveness to generate reimbursement coverage and payment specifically for the HeartBeam telehealth Generation 2 solution. We expect to be able to demonstrate significant clinical benefits for patients and savings to the health care system, justifying reimbursement levels well in excess of the amount paid through the RPM pathway.
For the telehealth Product, our primary marketing strategy will focus on the medical community with continued validation of clinical efficacy and cost-effectiveness and the establishment of reference sites. We plan to also create educational materials and provide other support to help educate our customers’ patients.
For the ED Product, the primary customers are acute care facilities. As with the telehealth Product, we plan to publish clinical studies on the effectiveness of the Product. In addition, we plan to develop financial models demonstrating the cost-effectiveness of the approach and establish reference sites who are using the Product. We do not expect to obtain specific reimbursement for the ED Product but intend to demonstrate that the purchase of the software would result in clinical and economic benefits and potentially reduced malpractice legal exposure for ED provider institutions.
We are establishing a direct sales network with relationships and experience selling to EDs.
Clinical Data
HeartBeam has performed three clinical studies to assess performance of our technologies.
1. HIDES — Included 66 patients whose coronary arteries were occluded during a Percutaneous Coronary Intervention (“PCI”) and had electrical signals simultaneously collected by both a traditional 12-lead ECG and our vector signal-based device.
ECG recordings:
66 baseline recordings for each patient were taken during patient enrollment and prior to the balloon inflation. These were ischemia negative recordings. 120 recordings were taken during balloon occlusions in various arteries. These were ischemia positive recordings. There were a total of 186 diagnostic recordings: 66 ischemia negative recordings and 120 ischemia positive recordings.
Study design:
The HeartBeam automated ischemia marker is based on the vector difference between ST vectors of the symptomatic and baseline recordings. Human reading results were obtained by averaging results from three expert readers (two electrophysiologists and one invasive cardiologist) who were presented with the 186 standard 12-lead ECG recordings. These readings were performed in two sessions four weeks apart. A total of six readings were averaged to arrive at human readers’ ischemia detection performance.
Study results:
The automated HeartBeam ischemia marker was superior to human expert reading for detecting acute ischemia (66 patients, 120 balloon occlusions; 186 total recordings) using ECG signals only:
| | | | | | | | | | | | | | | | | | | | |
| | SENSITIVITY [%] | | SPECIFICITY [%] | | ACCURACY [%] |
| Human readings | | 71.94 | | 70.96 | | 71.59 |
| HeartBeam ischemia marker | | 91.7 | | 95.5 | | 93 |
| | p<0.01 | | p<0.01 | | p<0.01 |
The ischemia marker showed an area under the curve (“AUC”) in the receiver operating characteristic (“ROC”) curve of 93.6%. HeartBeam’s marker accuracy was consistent all across the three culprit arteries (LAD, LCX, RCA), p=1.00. There was no statistically significant difference in accuracy between three human readers (p=1.00).
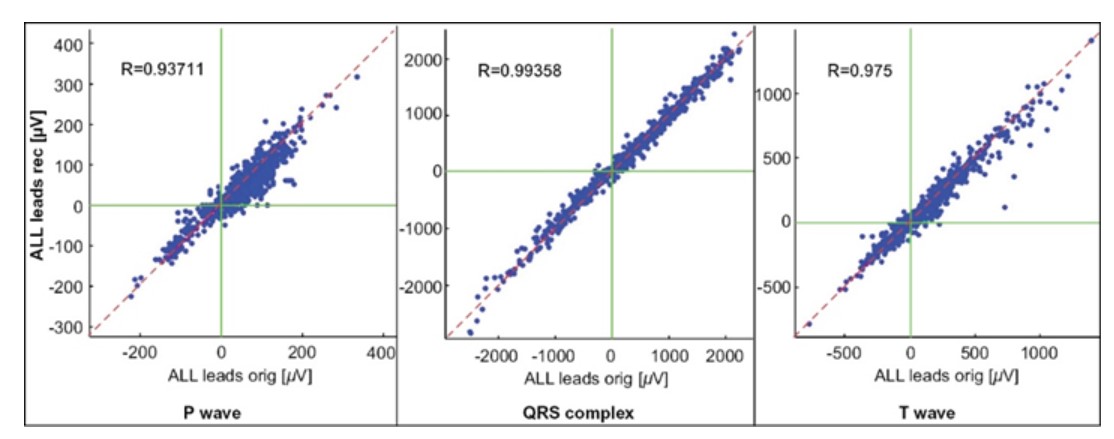
Synthetized vs, standard 12 lead ECG:
The synthesized 12 lead ECG was obtained using an individual transformation matrix obtained from standard 12 lead signals. Comparison of the standard 12 lead and synthetized 12 lead ECGs were performed with standard correlation analyses. Results for Pearson’s correlation coefficient R between standard 12 lead and synthetized 12 lead ECGs were:
P wave: R = 0.937;
QRS complex: R = 0.994;
T wave: R = 0.975
They are shown in the graphical form for all 186 pairs of signals (standard and synthesized 12-lead):
Pearson’s correlation coefficient is the test statistic that measures the statistical relationship between two continuous variables A value of 1 implies that a linear equation describes the relationship perfectly; value of 0 implies that there is no linear correlation between the variables
2. B Score — Evaluation of sensitivity and specificity of the HeartBeam diagnostic algorithm in diagnosing patients with ACS in the ED setting.
Study design and enrollment:
Enrolled were all patients presenting to an ED with chest pain or other symptoms suggestive of ACS who a) answered questions about risk factors and chest pain characteristics and b) had standard 12-lead ECG and HeartBeam ECG recorded 3 times with 10-15 minutes intervals between recordings. The final decision whether a patient was having ACS was determined by 3 cardiology experts (gold standard panel) based on discharge diagnosis and one week follow up data.
An additional HeartBeam ECG recording was taken between 9 and 12 months after the initial visit, when ST resolution was completed in the majority of cases, and this recording was used as the baseline ECG in HeartBeam’s diagnostic algorithm. The same set of data — risk factors, chest pain characteristics and three recordings — were both used for evaluation of the HeartBeam algorithm and presented to three expert cardiologists (evaluation panel) for a blinded evaluation.
Study results:
110 ER patients presenting with chest pain of which 29 (26%) with ACS (per gold standard panel), underwent HeartBeam assessment as well as assessment by the evaluation cardiologist panel. Sensitivity of the HeartBeam algorithm was 97% (27/28) and specificity was 56% (45/81). The only positive patient missed by the algorithm had known coronary disease with typical anginal episode resulting in a troponin leak. The average sensitivity and specificity of the evaluation cardiologists panel was 93% and 49%, respectively. The diagnostic performance of the HeartBeam algorithm in determining presence of the ACS in these patients was statistically indifferent from the performance of the evaluation cardiologist panel (p>0.42).
This result indicates that the quality of the advice produced by our expert system will be extremely valuable to the physician who is assessing the condition of a patient in a telehealth environment.
3. ISPEC — evaluation of the ECG signal quality and specificity of the ACS detection (false positive rate) in real-life use of the HeartBeam device by non-symptomatic patients.
Study design and enrollment:
The study included 30 participants, healthy volunteers and patients with different cardiac disturbances. The participants recorded three HeartBeam recordings three times daily, for 3 to 7 days. The ECG signal quality was evaluated as the percentage of recordings rejected by the HeartBeam cloud-based software due to insufficient signal quality. The specificity of HeartBeam in classification of non-ischemic recordings was performed by application of HeartBeam’s ACS detection algorithm. It was assumed that none of the patients was ischemic during the study period, as none reported chest pain symptoms.
Study results:
The study generated a total of 1845 recordings; 15.2% (282/1845) of the recordings were rejected by HeartBeam due to insufficient signal quality and had to be repeated. The false positive rate of the HeartBeam algorithm in classification of non-ischemic recordings was zero. In other words, the specificity was 100% in real-life use of our system in asymptomatic patients.
Competition
The cardiac monitoring and detection market is characterized by rapid technological change and strong competition. There are numerous companies developing technologies that are competitive, in a broad sense, to our products, and many of these companies have significantly greater resources than we do.
In the category of ambulatory (telehealth) cardiac monitors — devices that are intended to be used outside of a health facility setting — there are two major segments: consumer devices and devices prescribed for ACS.
Consumer Devices
The consumer device segment consists of devices that are FDA cleared but are sold directly to patients, without a prescription. Generally, these devices are single lead ECG devices intended to recognize heart rhythm abnormalities, such as atrial fibrillation, but are not intended for ischemia detection or for life threatening conditions such as heart attack.
•Apple Inc, a public company located in Cupertino, CA, produces the Apple Watch, which includes ECG functionality. The Apple Watch is a single lead ECG with two electrodes that contact the wrist and the finger and is intended to detect only Atrial Fibrillation.
•AliveCor Inc, a private company located in Mountain View, CA, produces the KardiaMobile, KardiaMobile Card and KardiaMobile 6L devices. These devices are intended to detect heart rhythm irregularities, such as Atrial Fibrillation, only.
•Google Inc, a public company located in Mountain View, CA, produces the Fitbit Sense smartwatch and ECG app. The Fitbit Sense watch is a single lead ECG with two electrodes that contact the fingers and is intended to detect only Atrial Fibrillation.
•Samsung Electronics Co., Ltd, based in Seoul, South Korea, is publicly traded in Korea. It produces the Galaxy Watch3 and Galaxy Watch Active2 smartwatches with ECG functionality, intended to detect only Atrial Fibrillation.
Devices prescribed for ischemia detection
There are a small number of devices that have been cleared by FDA to be used outside of healthcare facilities that provide information for patients with potential ischemic events such as MIs.
•Angel Medical Systems, Inc. is a private company based in Eatontown, NJ. The AngelMed Guardian is an implantable cardiac monitor for patients who are deemed to be extremely high risk for an MI. Physicians implant the AngelMed Guardian in patients. The company believes that the HeartBeam device will be a viable alternative to the AngelMed Guardian, as it does not require an implant and does not have a high up-front cost.
•SHL Telemedicine Ltd., is based in Tel Aviv, Israel and is publicly traded. It produces Smartheart Pro, a 12 lead ECG indicated for patient use at home. Smartheart Pro is larger and more complex than our telehealth solution, requiring the placement of an electrode belt, two underarm electrodes and a waist electrode, and moistening the areas before use. Most patients would find this technology not suited to be carried with them at all times because of the large size and complex lead attachment procedure.
There are several competitors in the category of software that automatically analyzes 12 lead ECGs performed in healthcare facilities, specifically in an ED. Major competitors in this market include the following:
•General Electric, a publicly traded company based in Chicago, IL produces a line of ECG equipment. The company also has developed the GE Marquette 12SL ECG analysis program, which analyzes the ST segment of the ECG to detect potential cardiac ischemia. It does not use the 3D vector approach in deriving a diagnostic suggestion.
•Koninklijke Philips N.V., a publicly traded company based in Amsterdam, NL, produces a range of ECG products, including products that feature the DXL algorithm for resting ECGs. The Philips DXL algorithm monitors the ST segment to detect STEMI. It does use the 3D vector approach in deriving a diagnostic suggestion.
Intellectual Property
We believe our innovations are protected with a strong patent portfolio. For a limited number of aspects of our proprietary technology we rely on trade secret protection. It is our view that the combination of these two methods of intellectual property protection maximizes our chances for success.
The issued and pending U.S. patent applications cover compact systems for remote detection and/or diagnosis of AMI. The pending EU and CN patent applications correspond to the pending and issued US cases. The pending PCT applications cover methods and apparatuses for automatic cardiac diagnosis as well as compact systems including retractable electrodes.
HeartBeam has three issued U.S. patents (U.S.10,433,744, U.S.10,117,592 and U.S.11,071,490), and six pending U.S applications. Three of the pending applications have published, all of the remaining three pending cases are unpublished. Outside of the U.S., HeartBeam has three pending EU utility patent applications (one of which is allowed), one pending CA utility patent application, one pending CN utility patent application, one pending JP utility patent application, one pending AU application, and two pending PCT applications. The issued patents are predicted to expire between April 11, 2036 and October 5, 2041.
The following table sets forth a brief description of issued and pending patents, including their respective titles:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Patent Type | | Application No. Pat. No. | | Status | | Predicted Expiration | | Title Summary |
Utility
(US) | | 15/096,159
US 10,433,744 | | Issued | | Sep 15, 2036 | | MOBILE THREE-LEAD CARDIAC MONITORING DEVICE AND METHOD FOR AUTOMATED DIAGNOSTICS
Methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(US) | | 15/632,155
US 10,117,592 | | Issued | | Apr 11, 2036 | | MOBILE THREE-LEAD CARDIAC MONITORING DEVICE AND METHOD FOR AUTOMATED DIAGNOSTICS
Methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(US) | | 17/092,152 | | Published | | Apr 11, 2036 | | MOBILE THREE-LEAD CARDIAC MONITORING DEVICE AND METHOD FOR AUTOMATED DIAGNOSTICS
Methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(US) | | 17/202,299
US 11,071,490 | | Issued | | Apr 11, 2036 | | ELECTROCARDIOGRAM PATCH DEVICES AND METHODS
Methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(CN) | | 201680030550.5 | | Published | | Apr 11, 2036 | | MOBILE THREE-LEAD CARDIAC MONITORING DEVICE AND METHOD FOR AUTOMATED DIAGNOSTICS
Methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(EU) | | 16777474.4 | | Allowed | | Apr 11, 2036 | | MOBILE THREE-LEAD CARDIAC MONITORING DEVICE AND METHOD FOR AUTOMATED DIAGNOSTICS
Methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(EU) | | 198948150 | | Pending | | Nov 18, 2039 | | HAND HELD DEVICE FOR AUTOMATIC CARDIAC RISK AND DIAGNOSTIC ASSESSMENT
Method and apparatus for performing automatic cardiac diagnosis. |
Utility
(US) | | 17/296,669 | | Pending | | Nov 18, 2039 | | HAND HELD DEVICE FOR AUTOMATIC CARDIAC RISK AND DIAGNOSTIC ASSESSMENT
Method and apparatus for performing automatic cardiac diagnosis. |
Utility
(US) | | 17/443,456 | | Pending | | Apr 11, 2036 | | ELECTROCARDIOGRAM PATCH DEVICES AND METHODS Adhesive patch methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(US) | | 17/570,368 | | Pending | | Apr 11, 2036 | | ELECTROCARDIOGRAM PATCH DEVICES AND METHODS
Adhesive patch methods and apparatuses for remote and detection and/or diagnosis of acute myocardial infarction (AMI). |
Utility
(US) | | 17/609,014 | | Pending | | May 20, 2040 | | COMPACT MOBILE THREE-LEAD CARDIAC MONITORING DEVICE
Compact, mobile three-lead cardiac monitoring devices for remote detection and/or diagnosis of cardiac events. |
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Patent Type | | Application No. Pat. No. | | Status | | Predicted Expiration | | Title Summary |
Utility
(AU) | | 2020275409 | | Pending | | May 20, 2040 | | COMPACT MOBILE THREE-LEAD CARDIAC MONITORING DEVICE
Compact, mobile three-lead cardiac monitoring devices for remote detection and/or diagnosis of cardiac events. |
Utility
(CA) | | 3137669 | | Pending | | May 20, 2040 | | COMPACT MOBILE THREE-LEAD CARDIAC MONITORING DEVICE Compact, mobile three-lead cardiac monitoring devices for remote detection and/or diagnosis of cardiac events. |
Utility
(EU) | | 208063123 | | Pending | | May 20, 2040 | | COMPACT MOBILE THREE-LEAD CARDIAC MONITORING DEVICE Compact, mobile three-lead cardiac monitoring devices for remote detection and/or diagnosis of cardiac events. |
Utility
(JP) | | 2021568329 | | Pending | | May 20, 2040 | | COMPACT MOBILE THREE-LEAD CARDIAC MONITORING DEVICE Compact, mobile three-lead cardiac monitoring devices for remote detection and/or diagnosis of cardiac events. |
Utility
(PCT) | | PCTUS2021
059271 | | Pending | | N/A | | COMPACT MOBILE THREE-LEAD CARDIAC MONITORING DEVICE WITH HYBRID ELECTRODE Compact, mobile three-lead cardiac monitoring devices for remote detection and/or diagnosis of cardiac events. |
Utility
(PCT) | | PCTUS2022
011075 | | Pending | | N/A | | AMBULATORY ELECTROCARDIOGRAM PATCH DEVICES AND METHODS Cardiac monitoring patch devices (e.g., an ECG patch for 12-lead detection) for remote detection and/or diagnosis of cardiac events (e.g., acute myocardial infarction). |
Utility
(US) | | 17/494,806 | | Pending | | Oct 5, 2041 | | METHOD AND APPARATUS FOR RECONSTRUCTING ELECTROCARDIOGRAM (ECG) DATA Synthesizing (generating) 12-lead ECG dataset from 3-lead ECG data. |
We have entered into, and generally plan to continue to enter into, non-disclosure, confidentiality and intellectual property assignment agreements with all new employees as a condition of employment. In addition, we intend to generally enter into confidentiality and non-disclosure agreements with consultants, manufacturers’ representatives, distributors, suppliers and others to attempt to limit access to, use and disclosure of our proprietary information. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
The ownership of all filed patents is assigned to HeartBeam, Inc. At this time one additional patent application is being drafted by us.
Research and Development
The primary objective of our research and development program is to provide innovative, user friendly solutions with high medical value. To date, we have been highly successful in developing our initial products. The emphasis has been on developing a user-friendly solution that is always with the patient and that provides assistance to physicians in diagnosing heart attacks in chest pain patients.
Our Research and Development team is largely based in Belgrade, Serbia. We have assembled a highly capable team currently consisting of PhD level contributors who are credited with developing our key inventions and patents. The diverse group includes nuclear physicists, signal processing specialists, and biomedical engineers. We plan to continue to utilize this team in the future.
Future research and development efforts are expected to focus on the application of signal processing and artificial intelligence to address a range of cardiac conditions.
Future Products
Our core technology — the heart vector approach adopted and invented by our scientific team — is a platform technology that can provide diagnostic solutions to a variety of cardiovascular patients. Our plans call for expanding solutions that diagnose all major cardiac conditions that are diagnosed by ECGs.
Our future plans include the development of a 12-lead capable patch ECG monitor that is expected to provide advantages over existing single-lead ECG patch products such as the Zio-XT from iRhythm Technologies, Inc. Our plan is to offer a 12-lead ECG with a patch that is very similar, in dimensions and look and feel, to the currently available single lead ECG patches. We believe providing standard of care 12-lead ECG capabilities will have significant diagnostic advantages over a single lead patch.
While our initial telehealth Product is powered by a software expert system that serves as a diagnostic aid to a physician, we plan to develop an AI based diagnostic system to supplement our diagnostic expert system.
Government Regulation
General
Our proposed products are subject to regulation by the U.S. Food and Drug Administration (“FDA”) and various other federal and state agencies, as well as by foreign governmental agencies. These agencies enforce laws and regulations that govern the development, testing, manufacturing, labeling, advertising, marketing and distribution, and market surveillance of our medical device products.
In addition to those indicated below, the only other regulations we encounter are regulations that are common to all businesses, such as employment legislation, implied warranty laws, and environmental, health and safety standards, to the extent applicable. We will also encounter in the future industry-specific government regulations that would govern our products, if and when developed for commercial use. It may become the case that other regulatory approvals will be required for the design and manufacture of our products and proposed products.
U.S. Regulation
The FDA governs the following activities that HeartBeam performs, will perform, upon the clearance or approval of its Product, or that are performed on its behalf, to ensure that medical products distributed domestically or exported internationally are safe and effective for their intended uses:
•product design, and development
•product safety, testing, labeling and storage
•record keeping procedures; and
•product marketing.
There are numerous FDA regulatory requirements governing the approval or clearance and subsequent commercial marketing of our products. These include:
•the timely submission of product listing and establishment registration information, along with associated establishment user fees;
•continued compliance with the Quality System Regulation, or QSR, which require specification developers and manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process;
•labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or off-label use or indication;
•clearance or approval of product modifications that could significantly affect the safety or effectiveness of the device or that would constitute a major change in intended use;
•Medical Device Reporting regulations (“MDR”), which require that manufacturers keep detailed records of investigations or complaints against their devices and to report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur;
•adequate use of the Corrective and Preventive Actions process to identify and correct or prevent significant systemic failures of products or processes or in trends which suggest same;
•post-approval restrictions or conditions, including post-approval study commitments;
•post-market surveillance regulations, which apply when necessary, to protect the public health or to provide additional safety and effectiveness data for the device; and
•notices of correction or removal and recall regulations.
Depending on the classification of the device, before HeartBeam can commercially distribute medical devices in the United States, it has to obtain, either prior 510(k) clearance, 510(k) de-novo clearance or premarket approval (PMA), from the
FDA unless a respective exemption applies to the device under review by the FDA.
The FDA classifies medical devices into one of three classes based on the degree of risk associated with each medical device and the extent of regulatory controls needed to ensure the device’s safety and effectiveness:
•Class I medical devices, which are low risk and subject to only general controls (e.g., registration and listing, medical device labeling compliance, MDRs, Quality System Regulations, and prohibitions against adulteration and misbranding) and, in some cases, to the 510(k) premarket clearance requirements;
•Class II medical devices, which are moderate risk and generally require 510(k) or 510(k) de-novo premarket clearance before they may be commercially marketed in the United States as well as general controls and potentially special controls like performance standards or specific labeling requirements; and
•Class III medical devices, which are devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a predicate device. Class III medical devices generally require the submission and approval of a PMA supported by clinical trial data.
The custom software and hardware of our products, we believe, are classified as Class II that will run on a Class I platform. Class II medical devices are those for which general controls alone are insufficient to provide reasonable assurance of safety and effectiveness and there is sufficient information to establish special controls. Special controls can include performance standards, post-market surveillance, patient histories and FDA guidance documents. Premarket review and clearance by the FDA for these devices is generally accomplished through the 510(k). As part of the 510(k), the FDA may have required the following:
•Development of comprehensive product description and indications for use;
•Completion of extensive preclinical tests and preclinical animal studies, performed in accordance with the FDA’s Good Laboratory Practice (“GLP”) regulations;
•Comprehensive review of predicate devices and development of data supporting the new product’s substantial equivalence to one or more predicate devices; and
•If appropriate and required, certain types of clinical trials (IDE submission and approval may be required for conducting a clinical trial in the US).
Clinical trials involve use of the medical device on human subjects under the supervision of qualified investigators in accordance with current Good Clinical Practices (“GCPs”), including the requirement that all research subjects provide informed consent for their participation in the clinical study. A written protocol with predefined end points, an appropriate sample size and pre-determined patient inclusion and exclusion criteria, is required before initiating and conducting a clinical trial. All clinical investigations of devices to determine safety and effectiveness must be conducted in accordance with the FDA’s Investigational Device Exemption, or IDE, regulations that among other things, govern investigational device labeling, prohibit promotion of the investigational device, and specify recordkeeping, reporting and monitoring
responsibilities of study sponsors and study investigators. If the device presents a “significant risk,” as defined by the FDA, the agency requires the device sponsor to submit an IDE application, which must become effective prior to commencing human clinical trials. The IDE will automatically become effective 30 days after receipt by the FDA, unless the FDA denies the application or notifies the company that the investigation is on hold and may not begin. If the FDA determines that there are deficiencies or other concerns with an IDE that requires modification, the FDA may permit a clinical trial to proceed under a conditional approval. In addition, the study must be approved by, and conducted under the oversight of, an Institutional Review Board (IRB) for each clinical site. If the device presents a non-significant risk to the patient, a sponsor may begin the clinical trial after obtaining approval for the trial by one or more IRBs without separate approval from the FDA, but it must still follow abbreviated IDE requirements, such as monitoring the investigation, ensuring that the investigators obtain informed consent, and labeling and record-keeping requirements.
Given successful completion of all required testing, a detailed 510(k) premarket notification or 510(k) de-novo will be submitted to the FDA requesting clearance to market the product. This notification will include all relevant data from pertinent preclinical and clinical trials, together with detailed information relating to the product’s manufacturing controls and proposed labeling, and other relevant documentation.
A 510(k) clearance letter from the FDA authorizes commercial marketing of the device for one or more specific indications of use.
After 510(k) clearance, HeartBeam is required to comply with a number of post-clearance requirements, including, but not limited to, Medical Device Reporting and complaint handling, and, if applicable, reporting of corrective actions. Also, quality control and manufacturing procedures must continue to conform to QSRs. The FDA periodically inspects manufacturing facilities to assess compliance with FDA’s Quality System Regulations (QSRs), which impose extensive procedural, substantive, and record keeping requirements on medical device manufacturers. In addition, changes to the manufacturing process are strictly regulated, and, depending on the change, validation activities may need to be performed. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with QSRs and other types of regulatory controls.
After a device receives 510(k) clearance from the FDA, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use or technological characteristics, requires a new 510(k) clearance or could require a PMA. The FDA requires each manufacturer to make the determination of whether a modification requires a new 510(k) notification or PMA in the first instance, but the FDA can review any such decision. If the FDA disagrees with a manufacturer’s decision not to seek a new 510(k) clearance or PMA for a particular change, the FDA may retroactively require the manufacturer to seek 510(k) clearance or PMA. The FDA can also require the manufacturer to cease U.S. marketing and/or recall the modified device until additional 510(k) clearance or PMA approval is obtained.
The FDA and the Federal Trade Commission, or FTC, will also regulate the advertising claims of HeartBeam’s products to ensure that the claims it makes are consistent with its regulatory clearances, that there is scientific data to substantiate the claims and that product advertising is neither false nor misleading.
We will apply for 510(k) clearance for both our cloud-based diagnostic engine for the ED product and the cloud-based diagnostic engine and hardware components of our telehealth Product. To obtain 510(k) clearance, a company must submit a notification to the FDA demonstrating that its proposed device is substantially equivalent to a predicate device (i.e., a device that was in commercial distribution before May 28, 1976, a device that has been reclassified from Class III to Class I or Class II, or a 510(k)-cleared device). The FDA’s 510(k) clearance process generally takes from three to 12 months from the date the application is submitted but also can take significantly longer. If the FDA determines that the device or its intended use is not substantially equivalent to a predicate device, the device is automatically placed into Class III, requiring the submission of a PMA. Once the information is submitted, there is no guarantee that the FDA will grant a company 510(k) clearance for its pipeline products, and failure to obtain the necessary clearances for its products would adversely affect its ability to grow its business. Delays in receipt or failure to receive the necessary clearances, or the failure to comply with existing or future regulatory requirements, could reduce its business prospects.
Devices that cannot be cleared through the 510(k) process due to lack of a predicate device but would be considered low or moderate risk may be eligible for the 510(k) de-novo process. In 1997, the Food and Drug Administration Modernization Act (“FDAMA”) added the de novo classification pathway now codified in section 513(f)(2) of the FD&C Act. This law established an alternate pathway to classify new devices into Class I or II that had automatically been placed in Class III
after receiving a Not Substantially Equivalent, or NSE, determination in response to a 510(k) submission. Through this regulatory process, a sponsor who receives an NSE determination may, within 30 days of receipt, request FDA to make a risk-based classification of the device through what is called a “de novo request.” In 2012, section 513(f)(2) of the FD&C Act was amended by section 607 of the Food and Drug Administration Safety and Innovation Act (“FDASIA”), in order to provide a second option for de novo classification. Under this second pathway, a sponsor who determines that there is no legally marketed device upon which to base a determination of substantial equivalence can submit a de novo request to FDA without first submitting a 510(k).
In the event that a company receives a Not Substantially Equivalent determination in response to a 510(k) submission, the device may still be eligible for the 510(k) de novo classification process.
Devices that cannot be cleared through the 510(k) or 510(k) de novo classification process require the submission of a PMA. The PMA process is much more time consuming and demanding than the 510(k) notification process. A PMA must be supported by extensive data, including but not limited to data obtained from preclinical and/or clinical studies and data relating to manufacturing and labeling, to demonstrate to the FDA’s satisfaction the safety and effectiveness of the device. After a PMA application is submitted, the FDA’s in-depth review of the information generally takes between one and three years and may take significantly longer.
We also need to establish a suitable and effective quality management system, which establishes controlled processes for our product design, manufacturing, and distribution. We plan to do this in compliance with the internationally recognized standard ISO 13485:2013 Medical Devices — Quality Management Systems — Requirements for Regulatory Purposes. Following the introduction of a product, the FDA and foreign agencies engage in periodic reviews of our quality systems, as well as product performance and advertising and promotional materials. These regulatory controls, as well as any changes in FDA policies, can affect the time and cost associated with the development, introduction and continued availability of new products. Where possible, we anticipate these factors in our product development processes. These agencies possess the authority to take various administrative and legal actions against us, such as product recalls, product seizures and other civil and criminal sanctions.
In order to reduce time and minimize the need to hire permanent technical and regulatory staffing in pursuing our FDA clearances we have contracted with a service provider to prepare our Products for FDA submission. This is the most efficient way to maximize our chances for timely FDA clearances.
Based on all available data and opinions from our well qualified external consultants who specialize in FDA submissions, we believe that both our initial products and the follow-on products qualify for the 510(k) clearance path.
Foreign Regulation
As we plan to market our products in the EU and other foreign markets, in addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products in foreign countries. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Preparations for FDA Clearance Submissions and Design for Manufacturing
Up to this point, we have focused primarily on research and development of our first two Products. At this time, we have fully functional versions of our first two Products which we have used in most of our medical studies. We consider them to be pre-production versions as they need to go through a full FDA class improvement and testing cycle before they can be presented to the FDA for a 510(k) clearance. We are not yet at a stage to commence volume production of our products.
We have a contract with LIVMOR to prepare our ED Product for clearance via a 510(K) submission to the FDA. The principal terms of this contract are to incorporate the product requirements, user needs, and 3D vector algorithm plugin developed by HeartBeam into the ED MI software product. Our contract with Triple Ring, Inc., US-based medical device and design organization (“TRIPLE RING”) as the development partner is to turn the ECG Hardware prototype into a commercially ready product cleared by the FDA. The principal terms of the agreement require TRIPLE RING to perform
the design, development, and testing of the ECG device and interface with the user application. With oversight from and in partnership with HeartBeam, TRIPLE RING will define all requirements, testing, detailed design, verification protocols, and test cases for all subsystems included in the ECG device product. Once the subsystems are complete, the service provider will perform system-level software testing. Once completed, the product will be provided to HeartBeam to perform system validation testing using clinical data from post-MI patients.
We plan to have a scalable manufacturing strategy.
Based on our extensive research and reference checking, we believe that TRIPLE RING, an FDA Registered and ISO 13485 Certified organization, has experience in both product development of medical devices and manufacturing capabilities that we intend to use in the limited market release of our telehealth Generation 1 Product. We plan to consider other manufacturers, inside and outside the US, for our high-volume manufacturing.
Employees
As of December 31, 2021, we had 5 full-time employees. We have budgeted to hire additional full-time employees (including additional consultants or independent contractors) in the near future to execute our growth plans.
We consider our employee relations to be good.
Corporate Information
Our principal executive offices are located at 2118 Walsh Avenue, Suite 200, Santa Clara, CA 95050. Our telephone number is (408) 899-4443 and our web address is www.heartbeam.com. Financial and other information can be accessed on the “Investors” section of our website. We make available through our website, free of charge, copies of our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act as soon as reasonably practicable after filing such material electronically or otherwise furnishing it to the Securities and Exchange Commission (the “SEC”). Also posted on our website are certain corporate governance documents, including our Code of Business Conduct and Ethics. The reference to our website is textual in reference only, and the information included or referred to on, or accessible through, our website does not constitute part of, and is not incorporated by reference into, this report or any other filing.
We also file periodic reports, proxy statements and other information with the SEC. Such reports may be obtained by visiting the Public Reference Room of the SEC at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling the SEC at (800) SEC-0330. In addition, the SEC maintains an internet site at http://www.sec.gov that contains reports, proxy and information statements and other information.
Item 1A. Risk Factors.
You should consider carefully the risks, uncertainties and other factors described below, in addition to the other information set forth in this Form 10-K, before making an investment decision. Any of these risks, uncertainties and other factors could materially and adversely affect our business, financial condition, results of operations, cash flows or prospects. In that case, the market price of our common stock could decline, and you may lose all or part of your investment in our common stock. See also “Cautionary Statement Regarding Forward-Looking Statements.”
Risks Related to Our Business
We have a limited operating history upon which investors can evaluate our future prospects.
We have a limited operating history upon which an evaluation of our business plan or performance and prospects can be made. The business and prospects of the Company must be considered in the light of the potential problems, delays, uncertainties and complications encountered in connection with a newly established business and new industry. The risks include, but are not limited to, the possibility that we will not be able to develop functional and scalable products and services, or that although functional and scalable, our products and services will not be economical to market; that our competitors hold proprietary rights that preclude us from marketing such products; that our competitors market a superior or equivalent product; that we are not able to upgrade and enhance our technologies and products to accommodate new features and expanded service offerings; or the failure to receive necessary regulatory clearances for our products. To successfully introduce and market our products at a profit, we must establish brand name recognition and competitive
advantages for our products. There are no assurances that we can successfully address these challenges and if unsuccessful, we and our business, financial condition and operating results could be materially and adversely affected.
The current and future expense levels of our business are based largely on estimates of planned operations and future revenues rather than experience. It is difficult to accurately forecast future revenues because our business is new and our market has not been developed. If our forecasts prove incorrect, the business, operating results and financial condition of the Company may be materially and adversely affected. Moreover, we may be unable to adjust our spending in a timely manner to compensate for any unanticipated reduction in revenues. As a result, any significant reduction in planned or actual revenues may immediately and adversely affect our business, financial condition and operating results.
We may need to raise additional capital to expand our business to meet our long-term business objectives. We have no revenues and we cannot predict when we will achieve first revenues and sustained profitability.
We have no revenues and cannot definitely predict when we will achieve revenues and profitability. We do not anticipate generating significant revenues until we successfully develop, achieve regulatory clearance, commercialize and sell our proposed products, of which we can give no assurance. We are unable to determine when we will generate significant revenues from the sale of any such products.
We cannot predict when we will achieve profitability, if ever. Our inability to become profitable may force us to curtail or temporarily discontinue our research and development programs and our day-to-day operations. Furthermore, there can be no assurance that profitability, if achieved, can be sustained on an ongoing basis.
We may never complete the development and commercialization of products that we are currently developing and future development of new generations of any of our other proposed products.
We have no assurance of success as to the completion and of the commercial launch of our products or the completion and development of any new generations of products that are currently under development or other proposed or contemplated products, for any of our target markets. We continue to seek to improve our technologies while we are developing them so that they result in commercially viable products. Failure to improve on any of our technologies could delay or prevent their successful development for our target markets. Developing any technology into a marketable product is a risky, time consuming and expensive process. You should anticipate that we will encounter setbacks, discrepancies requiring time consuming and costly redesigns and changes, and that there is the possibility of outright failure.
We may not meet our product development and commercialization milestones.
We have established milestones, based upon our expectations regarding our technologies, which we use to assess our progress toward developing our products. These milestones relate to technology development and design improvements as well as dates for achieving development goals. If our products exhibit technical defects or are unable to meet cost or performance goals, our commercialization schedule could be delayed and potential purchasers of our initial commercial products may decline to purchase such products or may opt to pursue alternative products.
We may also experience shortages of the components used in our devices. The contract manufacturing operations that we will use could be disrupted by fire, earthquake or other natural disaster, a labor-related disruption, failure in supply or other logistical channels, electrical outages or other reasons. If there were a disruption to manufacturing facilities, we would be unable to manufacture devices until these manufacturing capabilities are restored or alternative manufacturing facilities are engaged.
Generally, we have met our milestone schedules when making technological advances in our product. We can give no assurance that our development and commercialization schedule will continue to be met as we further develop products currently under development or any of our other future products.
Our business is dependent upon physicians utilizing and prescribing our solution; if we fail to engage physicians to utilize our solution, our revenues may never materialize or may not meet our projections.
The success of our cardiac diagnosis and monitoring business is dependent upon physicians prescribing and utilizing our solution. The utilization of our solution by physicians for use in the prescription of cardiac monitoring is directly influenced by a number of factors, including:
•the ability of the physicians with whom we work to obtain sufficient reimbursement and be paid in a timely manner for the professional services they provide in connection with the use of our monitoring solutions;
•establishing ourselves as a cardiac monitoring technology company by publishing peer reviewed publications showing efficacy of our solutions,
•our ability to educate physicians regarding the benefits of our cardiac monitoring solutions over alternative diagnostic monitoring solutions,
•our demonstrating that our proposed products are reliable and supported by us in the field;
•supplying and servicing sufficient quantities of products directly or through marketing alliances; and
•pricing our devices and technology service fees in a medical device industry that is becoming increasingly price sensitive.
If we are unable to drive physician utilization, our revenues may never materialize or may not meet our projections.
We are subject to extensive governmental regulations relating to the manufacturing, labeling and marketing of our products.
Our medical technology products and operations are subject to regulation by the FDA, and other foreign and local governmental authorities. These agencies enforce laws and regulations that govern the development, testing, manufacturing, labeling, advertising, marketing and distribution, and market surveillance of our medical products.
Under the United States Federal Food, Drug, and Cosmetic Act, medical devices are classified into one of three classes — Class I, Class II or Class III — depending on the degree of risk associated with each medical device and the extent of control needed to ensure safety and effectiveness. We believe that our products currently under development and planned products will be Class II medical devices. Class II medical devices are subject to additional controls, including full applicability of the Quality System Regulations, and requirements for 510(k) pre-market notification.
The FDA may disagree with the classification of a new Class II medical device and require the manufacturer of that device to apply for approval as a Class III medical device. In the event that the FDA determines that our Class II medical Products should be classified as Class III medical devices, we could be precluded from marketing the devices for clinical use within the United States for a period of time, the length of which depends on the specific change in the classification. Reclassification of our Class II medical Products as Class III medical devices could significantly increase our regulatory costs, including the timing and expense associated with required clinical trials and other costs.
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products in foreign countries. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
The policies of the FDA and foreign regulatory authorities may change and additional government regulations may be enacted which could prevent or delay regulatory approval of our products and could also increase the cost of regulatory compliance. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
The FDA and non-U.S. regulatory authorities require that our products be manufactured according to rigorous standards. These regulatory requirements may significantly increase our production costs and may even prevent us from making our products in quantities sufficient to meet market demand. If we change our approved manufacturing process, the FDA may need to review the process before it may be used. Failure to comply with applicable regulatory requirements discussed could subject us to enforcement actions, including warning letters, fines, injunctions and civil penalties, recall or seizure of our products, operating restrictions, partial suspension or total shutdown of our production, and even criminal prosecution.
Federal, state and non-U.S. regulations regarding the manufacture and sale of medical devices are subject to future changes. The complexity, timeframes and costs associated with obtaining marketing clearances are unknown. Although we cannot predict the impact, if any, these changes might have on our business, the impact could be material.
Following the introduction of a product, these agencies will also periodically review our design and manufacturing processes and product performance. The process of complying with the applicable good manufacturing practices, adverse event reporting, clinical trial and other requirements can be costly and time consuming, and could delay or prevent the production, manufacturing or sale of our products. In addition, if we fail to comply with applicable regulatory requirements, it could result in fines, delays or suspensions of regulatory clearances, closure of manufacturing sites, seizures or recalls of products and damage to our reputation. Recent changes in enforcement practice by the FDA and other agencies have resulted in increased enforcement activity, which increases the compliance risk for the Company and other companies in our industry. In addition, governmental agencies may impose new requirements regarding registration, labeling or prohibited materials that may require us to modify or re-register products already on the market or otherwise impact our ability to market our products in those countries. Once clearance or approval has been obtained for a product, there is an obligation to ensure that all applicable FDA, and other regulatory requirements continue to be met.
Additionally, injuries caused by the malfunction or misuse of cardiac devices, even where such malfunction or misuse occurs with respect to one of our competitor’s products, could cause regulatory agencies to implement more conservative regulations on the medical cardiac monitoring industry, which could significantly increase our operating costs.
If we are not able to both obtain and maintain adequate levels of third-party reimbursement for our products, it would have a material adverse effect on our business.
Healthcare providers and related facilities are generally reimbursed for their services through payment systems managed by various governmental agencies worldwide, private insurance companies, and managed care organizations. The manner and level of reimbursement in any given case may depend on the site of care, the procedure(s) performed, the final patient diagnosis, the device(s) utilized, available budget, the efficacy, safety, performance and cost-effectiveness of our planned products and services, or a combination of these or other factors, and coverage and payment levels are determined at each payer’s discretion. The coverage policies and reimbursement levels of these third-party payers may impact the decisions of healthcare providers and facilities regarding which medical products they purchase and the prices they are willing to pay for those products. Thus, changes in reimbursement levels or methods may impact sales of our products.
We have no direct control over payer decision-making with respect to coverage and payment levels for our medical device products and services. Additionally, we expect many payers to continue to explore cost-containment strategies (e.g., comparative and cost- effectiveness studies, so-called “pay-for-performance” programs implemented by various public and private payers, and expansion of payment bundling schemes such as Accountable Care Organizations, and other such methods that shift medical cost risk to providers) that may potentially impact coverage and/or payment levels for our products and services.
The ability of physicians and other providers to successfully utilize our cardiac diagnostic and monitoring solutions and successfully allow payors to reimburse for the physicians’ technical and professional fees is critical to our business because physicians and their patients will select solutions other than ours in the event that payors refuse to adequately reimburse our technical fees and physicians’ professional fees.
Changes in reimbursement practices of third-party payers could affect the demand for our products and services and our revenue levels.
The sales of our proposed products and services could depend, in part, on the extent to which healthcare providers and facilities or individual users are reimbursed by government authorities, private insurers and other third-party payers for the costs of our products or the services performed with our products. The coverage policies and reimbursement levels of third-party payers, which can vary among public and private sources and by country, may affect which products customers purchase and the prices they are willing to pay for those products and services in a particular jurisdiction. Reimbursement rates can also affect the acceptance rate of new technologies. Legislative or administrative reforms to reimbursement systems in the United States or abroad, or changes in reimbursement rates by private payers, could significantly reduce reimbursement for medical actions using the Company’s products or result in denial of reimbursement for those products, which would adversely affect customer demand or the price customers may be willing to pay for such products and services.
We may experience difficulty in obtaining reimbursement for our services from commercial payors that consider our technology to be experimental and investigational, which would adversely affect our revenue and operating results.
Many commercial payors refuse to enter into contracts to reimburse the fees associated with medical devices or services that such payors determine to be “experimental and investigational.” Commercial payors typically label medical devices or services as “experimental and investigational” until such devices or services have demonstrated product superiority evidenced by a randomized clinical trial.
For example, clinical trials have been performed on some mobile cardiac telemetry devices, proving higher diagnostic yield than monitoring devices and services that are already being reimbursed. Certain remaining commercial payors, however, have stated that they do not believe the data from the clinical trials justifies the removal of the experimental designation for mobile cardiac telemetry solutions. As a result, certain commercial payors may refuse to reimburse the technical and professional fees associated with cardiac monitoring solutions such as the one expected to be offered by the Company.
If commercial payors decide not to reimburse physicians or providers for their services during the utilization of our cardiac monitoring solutions, our revenue could fail to materialize or meet our projections.
Reimbursement by Medicare is highly regulated and subject to change; our failure to comply with applicable regulations could decrease our expected revenue and may subject us to penalties or have an adverse impact on our business.
The Medicare program is administered by CMS, which imposes extensive and detailed requirements on medical services providers, including, but not limited to, rules that govern how we structure our relationships with physicians, and how and
where we provide our cardiac solutions. Our failure to comply with applicable Medicare rules could result in the inability of physicians to receive reimbursement as they will likely utilize our cardiac monitoring solution under the Medicare payment program.
Consolidation of commercial payors could result in payors eliminating coverage of mobile cardiac monitoring solutions or reducing reimbursement rates.
When payors combine their operations, the combined company may elect to reimburse physicians for cardiac monitoring services at the lowest rate paid by any of the participants in the consolidation. If one of the payors participating in the consolidation does not reimburse for these services at all, the combined company may elect not to reimburse at any rate. Reimbursement rates tend to be lower for larger payors. As a result, as payors consolidate, our expected average reimbursement rate may decline.
Product defects could adversely affect the results of our operations.
The design, manufacture and marketing of our hardware and software products involve certain inherent risks. Manufacturing or design defects, unanticipated use of our products, or inadequate disclosure of risks relating to the use of our products can lead to injury or other adverse events. These events could lead to recalls or safety alerts relating to our products (either voluntary or required by the FDA, or similar governmental authorities in other countries), and could result, in certain cases, in the removal of a product from the market. A recall could result in significant costs, as well as negative publicity and damage to our reputation that could reduce demand for our products. Personal injuries or deaths relating to the use of our products could also result in product liability claims being brought against us. In some circumstances, such adverse events could also cause delays in new product approvals.
Interruptions or delays in telecommunications systems or in the data services provided to us by cellular communication providers or the loss of our wireless or data services could impair the delivery of our cardiac monitoring services.
The success of our cardiac monitoring services will be dependent upon our ability to transmit, store, retrieve, process and manage data and to maintain and upgrade our data processing and communication capabilities. Our monitoring solution relies on a third-party wireless carrier to transmit data over its data network. All data sent by our monitors via this wireless data network is expected to be routed directly to healthcare providers and data centers or third-party ECG monitoring centers. We are therefore dependent upon a third party wireless carrier to provide data transmission services to us.
As we expand our commercial activities, an increased burden is expected to be placed upon our data processing systems and the equipment upon which they rely. Interruptions of our data networks, or the data networks of our wireless carrier, for any extended length of time, loss of stored data or other computer problems could have a material adverse effect on our business and operating results. Frequent or persistent interruptions in our cardiac monitoring services could cause permanent harm to our reputation and could cause current or potential users or prescribing physicians to believe that our systems are unreliable, leading them to switch to our competitors. Such interruptions could result in liability, claims and litigation against us for damages or injuries resulting from the disruption in service.
Our systems are also expected to be vulnerable to damage to or interruption of telecommunication services from earthquakes, floods, fires, power loss, telecommunication failures, terrorist attacks, computer viruses, break-ins, sabotage, and acts of vandalism. Despite any precautions that we may take, the occurrence of a natural disaster or other unanticipated problems could result in lengthy interruptions in these services. We do not carry business interruption insurance to protect against losses that may result from interruptions in service as a result of system failures. Moreover, the communications and information technology industries are subject to rapid and significant changes, and our ability to operate and compete is dependent on our ability to update and enhance the communication technologies used in our systems and services.
Interruptions in computing and data management cloud systems could impair the delivery of our cardiac monitoring services.
The success of our cardiac monitoring services will be dependent upon our ability to perform computing functions associated with our cardiac signal processing algorithms and data management. The diagnostic and monitoring functions rely on the uninterrupted availability of third-party cloud based computational and data management services. Availability of the cloud-based infrastructure is a critical link in our ability to deliver our services and could have a material adverse effect on our business and operating results. Furthermore, loss of data due to catastrophic events at the sites for these cloud based computer systems could cause permanent harm to our customers. These adverse events associated with unavailability of our cloud based computational infrastructure could result in liability, claims and litigation against us for damages or injuries resulting from the disruption in service.
Our systems are also expected to be vulnerable to damage or interruption in cloud computational services from earthquakes, floods, fires, power loss, technical failures, terrorist attacks, computer viruses, break-ins, sabotage, and acts of vandalism. Despite any precautions that we may take, the occurrence of a natural disaster or other unanticipated problems could result in lengthy interruptions in these services.
We could be exposed to significant liability claims if we are unable to obtain insurance at acceptable costs and adequate levels or otherwise protect ourselves against potential product liability claims.
The testing, manufacture, marketing and sale of medical devices entail the inherent risk of liability claims or product recalls. Product liability insurance is expensive and, if available, may not be available on acceptable terms at all periods of time. A successful product liability claim or product recall could inhibit or prevent the successful commercialization of our products, cause a significant financial burden on the Company, or both, which in either case could have a material adverse effect on our business and financial condition.
We require additional capital to support our present business plan and our anticipated business growth, and such capital may not be available on acceptable terms, or at all, which would adversely affect our ability to operate.
We will require additional funds to further develop our business plan. Based on our current operating plans, we plan to use $8.2 million in capital to fund our planned operations and sales efforts necessary to commercially launch our products. We plan to use the remainder of the $14.7 million net proceeds from our IPO for working capital and general corporate purposes. We may choose to raise additional capital beyond this in order to expedite and propel growth more rapidly. We can give no assurance that we will be successful in raising any additional funds. Additionally, if we are unable to generate sufficient planned revenues from our sales and operating activities, we may need to raise additional funds, doing so through debt and equity offerings, in order to meet our expected future liquidity and capital requirements, including capital required for the development completion and introduction of our future products and technologies. Any such financing that we undertake will likely be dilutive to current stockholders.
We intend to continue to make investments to support our business growth, including patent or other intellectual property asset creation. In addition, we may also need additional funds to respond to business opportunities and challenges, including our ongoing operating expenses, protecting our intellectual property, satisfying debt payment obligations, developing new lines of business and enhancing our operating infrastructure. While we may need to seek additional funding for such purposes, we may not be able to obtain financing on acceptable terms, or at all. In addition, the terms of our financings may be dilutive to, or otherwise adversely affect, holders of our Common Stock. We may also seek to raise additional funds through arrangements with collaborators or other third parties. We may not be able to negotiate any such arrangements on acceptable terms, if at all. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all our business plans.
We cannot predict our future capital needs and we may not be able to secure additional financing.
We will need to raise additional funds in the future to fund our working capital needs and to fund further expansion of our business. We may require additional equity or debt financings, collaborative arrangements with corporate partners or funds from other sources for these purposes. No assurance can be given that necessary funds will be available for us to finance our development on acceptable terms, if at all. Furthermore, such additional financings may involve substantial dilution of our stockholders or may require that we relinquish rights to certain of our technologies or products. In addition, we may experience operational difficulties and delays due to working capital restrictions. If adequate funds are not available from operations or additional sources of financing, we may have to delay or scale back our growth plans.
The results of our research and development efforts are uncertain and there can be no assurance of the commercial success of our products.
We believe that we will need to incur additional research and development expenditures to continue development of our existing proposed products as well as research and development expenditures to develop new products and services. The products and services we are developing and may develop in the future may not be technologically successful. In addition, the length of our product and service development cycle may be greater than we originally expected, and we may experience delays in product development. If our resulting products and services are not technologically successful, they may not achieve market acceptance or compete effectively with our competitors’ products and services.
If we fail to retain certain of our key personnel and attract and retain additional qualified personnel, we might not be able to pursue our growth strategy.
Our future success will depend upon the continued service of Dr. Branislav Vajdic and other members of our key management team and our technical contributors. Though no individual is indispensable, the loss of the services of these individuals could have a material adverse effect on our business, operations, revenues or prospects. We do not currently maintain key man life insurance on the lives of these individuals.
We will not be profitable unless we can demonstrate that our products can be manufactured at low prices.
To date, we have focused primarily on research and development of the first generation version of our software and hardware products, as well as other technologies we plan to introduce in our eco-system, and their proposed marketing and distribution. Consequently, we have little experience in manufacturing these products on a commercial basis. We plan to manufacture our products through third-party manufacturers. We can offer no assurance that either we or our
manufacturing partners will develop efficient, automated, low-cost manufacturing capabilities and processes to meet the quality, price, engineering, design and production standards or production volumes required to successfully mass market our products, especially at the low-cost levels we require to absorb the cost of near free distribution of our products pursuant to our proposed business plan. Even if we or our manufacturing partners are successful in developing such manufacturing capability and processes, we do not know whether we or they will be timely in meeting our product commercialization schedule or the production and delivery requirements of potential customers. A failure to develop such manufacturing processes and capabilities could have a material adverse effect on our business and financial results. If we or our suppliers fail to achieve or maintain regulatory approval of manufacturing facilities, our growth could be limited and our business could be harmed.
In order to maintain compliance with FDA and other regulatory requirements, our development and manufacturing facilities must be periodically re-evaluated and qualified under a quality system to ensure they meet production and quality standards. Suppliers of components and products used to manufacture our devices must also comply with FDA regulatory requirements, which often require significant resources and subject us and our suppliers to potential regulatory inspections and stoppages. If we or our suppliers do not maintain regulatory approval for our manufacturing operations, our business could be adversely affected.
Our dependence on a limited number of suppliers may prevent us from delivering our devices on a timely basis.
We currently rely on a limited number of suppliers of components for our prototype devices. If these suppliers became unable to provide components in the volumes needed or at an acceptable price, we would have to identify and qualify acceptable replacements from alternative sources of supply. The process of qualifying suppliers is lengthy. Delays or interruptions in the supply of our requirements could limit or stop our ability to provide sufficient quantities of devices on a timely basis or meet demand for our devices or services, which could have a material adverse effect on our business, financial condition and results of operations.
We rely significantly on information technology and any failure, inadequacy, or security lapse of that technology, including any cybersecurity incidents, could harm us.
We believe that companies have been increasingly subject to a wide variety of security incidents, cyberattacks and other attempts to gain unauthorized access. These threats can come from a variety of sources, ranging in sophistication from an individual hacker to a state-sponsored attack. Cyber threats may be generic, or they may be custom-crafted against our information systems. Over the past few years, cyber-attacks have become more prevalent and much harder to detect and defend against. Several key areas of our business depend on the use of information technologies, including production, manufacturing, marketing, and logistics, as well as clinical and regulatory matters. We also utilize systems that allow for the secure storage and transmission of proprietary or confidential information regarding our customers, employees, and others, including personal information. Despite our efforts to prevent such behavior, third parties may nonetheless attempt to hack into our systems and obtain data relating to our pre-clinical studies, clinical trials, patients using our VCG technology and our telehealth ECG collection device or other information relating to us or our business. If we fail to maintain or protect our information systems and data integrity effectively, we could have problems in determining product cost estimates and establishing appropriate pricing, have difficulty preventing, detecting, and controlling fraud, have disputes with physicians, and other health care professionals, have regulatory sanctions or penalties imposed, have increases in operating expenses, incur expenses or lose revenues as a result of a data privacy breach, or suffer other adverse consequences and reputational damages. While we have invested in the protection of data and information technology, there can be no assurance that our efforts or those of our third-party collaborators, if any, or manufacturers, to implement adequate security and quality measures for data processing would be sufficient to protect against data deterioration or loss in the event of a system malfunction, or to prevent data from being stolen or corrupted in the event of a security breach. Any such loss or breach could harm our business, operating results, and financial condition.
We have identified weaknesses in our internal controls, and we cannot provide assurances that these weaknesses will be effectively remediated or that additional material weaknesses will not occur in the future.
We do not yet have effective disclosure controls and procedures, or internal controls over all aspects of our financial reporting. We are continuing to develop and refine our disclosure controls and other procedures that are designed to ensure that information required to be disclosed by us in the reports that we will file with the SEC is recorded, processed, summarized and reported within the time periods specified in SEC rules and forms. Our management has deemed certain conditions to be material weaknesses and significant deficiencies in our internal controls. For example, we failed to employ a sufficient number of staff to maintain optimal segregation of duties and to provide optimal levels of oversight. Our management is responsible for establishing and maintaining adequate internal control over our financial reporting, as defined in Rule 13a-15(f) under the Exchange Act. We will be required to expend time and resources to further improve our internal controls over financial reporting, including by expanding our staff. However, we cannot assure you that our internal control over financial reporting, as modified, will enable us to identify or avoid material weaknesses in the future.
Our current controls and any new controls that we develop may become inadequate because of changes in conditions in our business, including increased complexity resulting from our international expansion. Further, weaknesses in our disclosure controls or our internal control over financial reporting may be discovered in the future. Any failure to develop or maintain effective controls, or any difficulties encountered in their implementation or improvement, could harm our operating results or cause us to fail to meet our reporting obligations and may result in a restatement of our financial statements for prior periods. Any failure to implement and maintain effective internal control over financial reporting could also adversely affect the results of management reports and independent registered public accounting firm audits of our internal control over financial reporting that we will eventually be required to include in our periodic reports that will be filed with the SEC. Ineffective disclosure controls and procedures, and internal control over financial reporting could also cause investors to lose confidence in our reported financial and other information, which would likely have a negative effect on the market price of our Common Stock.
We are not currently required to comply with the SEC rules that implement Section 404 of the Sarbanes-Oxley Act, and are therefore not required to make a formal assessment of the effectiveness of our internal control over financial reporting for that purpose. As a public company, we will be required to provide an annual management report on the effectiveness of our internal control over financial reporting commencing with our second annual report on Form 10-K. Our independent registered public accounting firm is not required to audit the effectiveness of our internal control over financial reporting until after we are no longer an “emerging growth company” as defined in the JOBS Act. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our internal control over financial reporting is documented, designed or operating.
Any failure to maintain effective disclosure controls and internal control over financial reporting could have a material and adverse effect on our business and operating results, and cause a decline in the market price of our Common Stock.
Risks Related to Economic Condition, Including as a result of the Covid-19 Pandemic
Escalating global trade tensions, and the conflict between Russia and Ukraine, and the adoption or expansion of tariffs and trade restrictions could negatively impact us.
The current military conflict between Russia and Ukraine could lead to disruption, instability and volatility in global markets and industries that could negatively impact our operations and could adversely affect our business and/or our supply chain, business partners or customers in other countries beyond Russia and Ukraine. The U.S. government and other governments in jurisdictions in which we operate have imposed severe sanctions and export controls against Russia and Russian interests and threatened additional sanctions and controls. It is not possible to predict the broader consequences of this conflict, which could include sanctions, embargoes, regional instability, geopolitical shifts and adverse effects on macroeconomic conditions, currency exchange rates and financial markets. The impact of these measures, as well as potential responses to them by Russia, is currently unknown and they could adversely affect our business, supply chain, partners or customers. More specifically, while it is difficult to anticipate the impact the sanctions announced to date may have on our Research and Development team is largely based in Belgrade, Serbia any further sanctions imposed or actions taken by the U.S. or other countries, and any retaliatory measures by Russia in response, such as restrictions on energy supplies from Russia to countries in the region, could increase our costs, reduce our sales and earnings or otherwise have an adverse effect on our operations.
Natural disasters and other events beyond our control could materially adversely affect us.
Natural disasters or other catastrophic events may cause damage or disruption to our operations, international commerce and the global economy, and thus could have a strong negative effect on us. Our business operations are subject to interruption by natural disasters, fire, power shortages, pandemics and other events beyond our control. Such events could make it difficult or impossible for us to deliver our products and services to our customers and could decrease demand for our products and services. The World Health Organization declared the COVID-19 outbreak a pandemic. The extent of the impact of COVID-19 on our operational and financial performance will depend on certain developments, including the duration and spread of the outbreak, the impact on our customers and employees, all of which are uncertain and cannot be predicted. At this point, the overall extent to which COVID-19 may impact our financial condition or results of operations is uncertain.
The ongoing COVID-19 pandemic continues to present operational, health, labor, logistics and other challenges, and it is difficult to assess the ultimate impact of the COVID-19 pandemic on our business, financial condition and cash flows.
There are many variables and uncertainties regarding the COVID -19 pandemic, including the emergence, contagiousness and threat of new and different strains of the virus and their severity; the effectiveness of treatments or vaccines against the virus or its new strains; the extent of travel restrictions, business closures and other measures that are or may be imposed in affected areas or countries by governmental authorities; disruptions in the supply chain; an increasingly competitive labor market due to a sustained labor shortage or increased turnover caused by the COVID -19 pandemic; increased logistics costs; additional costs due to remote working arrangements, adherence to social distancing guidelines and other COVID-19 related challenges. Further, there remain increased risks of cyberattacks on information technology systems used in remote working environment; increased privacy-related risks due to processing health-related personal information; absence of workforce due to illness; and other factors that are currently unknown or considered immaterial. It is difficult to assess the ultimate impact of the COVID -19 pandemic on our business, financial condition and cash flows.
Risks Related to Our Industry
The industry in which we operate is highly competitive and subject to rapid technological change. If our competitors are better able to develop and market products that are safer, more effective, less costly, easier to use, or are otherwise more attractive, we may be unable to compete effectively with other companies.
The medical technology industry is characterized by intense competition and rapid technological change, and we will face competition on the basis of product features, clinical outcomes, price, services and other factors. Competitors may include large medical device and other companies, some of which have significantly greater financial and marketing resources than we do, and firms that are more specialized than we are with respect to particular markets. Our competition may respond more quickly to new or emerging technologies, undertake more extensive marketing campaigns, have greater financial, marketing and other resources than ours or may be more successful in attracting potential customers, employees and strategic partners.
Our competitive position will depend on multiple, complex factors, including our ability to achieve regulatory clearance and market acceptance for our products, develop new products, implement production and marketing plans, secure regulatory approvals for products under development and protect our intellectual property. In some instances, competitors may also offer, or may attempt to develop, alternative systems that may be delivered without a medical device or with a medical device superior to ours. The development of new or improved products, processes or technologies by other companies may render our products or proposed products obsolete or less competitive. The entry into the market of manufacturers located in low-cost manufacturing locations may also create pricing pressure, particularly in developing markets. Our future success depends, among other things, upon our ability to compete effectively against current technology, as well as to respond effectively to technological advances or changing regulatory requirements, and upon our ability to successfully implement our marketing strategies and execute our research and development plan. Our research and development efforts are aimed, in part, at solving increasingly complex problems, as well as creating new technologies, and we do not expect that all of our projects will be successful. If our research and development efforts are unsuccessful, our future results of operations could be materially harmed.
We face competition from other medical device companies that focus on similar markets.
We face competition from other companies that have longer operating histories and may have greater name recognition and substantially greater financial, technical and marketing resources than us. Many of these companies also have FDA or other applicable governmental approval to market and sell their products, and more extensive customer bases, broader customer relationships and broader industry alliances than us, including relationships with many of our potential customers. Increased competition from any of these sources could result in our failure to achieve and maintain an adequate level of customers and market share to support the cost of our operations.
Unsuccessful clinical trials or procedures relating to products under development could have a material adverse effect on our prospects.
The regulatory approval process for new products and new indications for existing products requires extensive clinical trials and procedures, including early clinical experiences and regulatory studies. Unfavorable or inconsistent clinical data from current or future clinical trials or procedures conducted by us, our competitors, or third parties, or perceptions regarding this clinical data, could adversely affect our ability to obtain necessary approvals and the market’s view of our future prospects. Such clinical trials and procedures are inherently uncertain and there can be no assurance that these trials or procedures will be completed in a timely or cost- effective manner or result in a commercially viable product. Failure to successfully complete these trials or procedures in a timely and cost-effective manner could have a material adverse effect on our prospects. Clinical trials or procedures may experience significant setbacks even after earlier trials have shown promising results. Further, preliminary results from clinical trials or procedures may be contradicted by subsequent clinical analysis. In addition, results from our already completed clinical trials or procedures may not be supported by actual long-
term studies or clinical experience. If preliminary clinical results are later contradicted, or if initial results cannot be supported by actual long-term studies or clinical experience, our business could be adversely affected. Clinical trials or procedures may be suspended or terminated by us, the FDA or other regulatory authorities at any time if it is believed that the trial participants face unacceptable health risks.
Intellectual property litigation and infringement claims could cause us to incur significant expenses or prevent us from selling certain of our products.
The medical device industry in which we operate is characterized by extensive intellectual property litigation and, from time to time, we might be the subject of claims by third parties of potential infringement or misappropriation. Regardless of outcome, such claims are expensive to defend and divert the time and effort of our management and operating personnel from other business issues. A successful claim or claims of patent or other intellectual property infringement against us could result in our payment of significant monetary damages and/or royalty payments, or it could negatively impact our ability to sell current or future products in the affected category and could have a material adverse effect on business, cash flows, financial condition or results of operations.
If we are unable to protect the confidentiality of our trade secrets, our business and competitive position would be harmed.
In addition to obtaining intellectual property protections we rely on trade secrets, including unpatented know-how, technology and other proprietary information, to maintain our competitive position. We will seek to protect these trade secrets, in part, by entering into non-disclosure and confidentiality agreements with parties who have access to them, such as our employees, corporate collaborators, outside scientific collaborators, contract manufacturers, consultants, advisors and other third parties. We will seek to protect our confidential proprietary information, in part, by entering into confidentiality and invention or intellectual property assignment agreements with our employees and consultants. Moreover, to the extent we enter into such agreements, any of these parties may breach the agreements and disclose our proprietary information, including our trade secrets, and we may not be able to obtain adequate remedies for such breaches. Enforcing a claim that a party illegally disclosed or misappropriated a trade secret is difficult, expensive and time-consuming, and the outcome is unpredictable. In addition, some courts inside and outside the United States are less willing or unwilling to protect trade secrets. If any of our trade secrets were to be lawfully obtained or independently developed by a competitor, we would have no right to prevent them, or those to whom they communicate it, from using that technology or information to compete with us. If any of our trade secrets were to be disclosed to or independently developed by a competitor, our competitive position would be harmed. In general, any loss of trade secret protection or other unpatented proprietary rights could harm our business, results of operations and financial condition.
If we are unable to protect our proprietary rights, or if we infringe on the proprietary rights of others, our competitiveness and business prospects may be materially damaged.
We have filed for and were granted a number of utility patents in the U.S as well as through PCT covering international markets. We will continue to seek patent protection for our inventions and may seek patent protection for our proprietary designs if warranted. Seeking patent protection is a lengthy and costly process, and there can be no assurance that patents will be issued from any pending applications, or that any claims allowed from existing or pending patents will be sufficiently broad or strong to protect our designs or our proprietary technology. There is also no guarantee that any patents we hold will not be challenged, invalidated or circumvented, or that the patent rights granted will provide competitive advantages to us. Our competitors have developed and may continue to develop and obtain patents for technologies that are similar or superior to our technologies. In addition, the laws of foreign jurisdictions in which we develop, manufacture or sell our products may not protect our intellectual property rights to the same extent, as do the laws the United States.
Adverse outcomes in legal disputes regarding patent and other intellectual property rights could result in the loss of our intellectual property rights, subject us to significant liabilities to third parties, require us to seek licenses from third parties on terms that may not be reasonable or favorable to us, prevent us from manufacturing, importing or selling our products, or compel us to redesign our products to avoid infringing third parties’ intellectual property. As a result, we may be required to incur substantial costs to prosecute, enforce or defend our intellectual property rights if they are challenged. Any of these circumstances could have a material adverse effect on our business, financial condition and resources or results of operations.
Dependence on our proprietary rights and failing to protect such rights or to be successful in litigation related to such rights may result in our payment of significant monetary damages or impact offerings in our product portfolios.
Our long-term success largely depends on our ability to market technologically competitive products. If we fail to obtain or maintain adequate intellectual property protection, we may not be able to prevent third parties from using our proprietary technologies or may lose access to technologies critical to our products. Also, our currently pending industrial design patent or any future patents applications may not result in issued patents, and issued patents are subject to claims concerning priority, scope and other issues.
Furthermore, to the extent we do not file applications for patents domestically or internationally, we may not be able to prevent third parties from using our proprietary technologies or may lose access to technologies critical to our products in other countries.
Enforcement of federal and state laws regarding privacy and security of patient information may adversely affect our business, financial condition or operations.
The use and disclosure of certain health care information by health care providers and their business associates have come under increasing public scrutiny. Recent federal standards under the Health Insurance Portability and Accountability Act of 1996, or HIPAA, establish rules concerning how individually-identifiable health information may be used, disclosed and protected. Historically, state law has governed confidentiality issues, and HIPAA preserves these laws to the extent they are more protective of a patient’s privacy or provide the patient with more access to his or her health information. As a result of the implementation of the HIPAA regulations, many states are considering revisions to their existing laws and regulations that may or may not be more stringent or burdensome than the federal HIPAA provisions. We must operate our business in a manner that complies with all applicable laws, both federal and state, and that does not jeopardize the ability of our customers to comply with all applicable laws. We believe that our operations are consistent with these legal standards. Nevertheless, these laws and regulations present risks for health care providers and their business associates that provide services to patients in multiple states. Because these laws and regulations are recent, and few have been interpreted by government regulators or courts, our interpretations of these laws and regulations may be incorrect. If a challenge to our activities is successful, it could have an adverse effect on our operations, may require us to forego relationships with customers in certain states and may restrict the territory available to us to expand our business. In addition, even if our interpretations of HIPAA and other federal and state laws and regulations are correct, we could be held liable for unauthorized uses or disclosures of patient information as a result of inadequate systems and controls to protect this information or as a result of the theft of information by unauthorized computer programmers who penetrate our network security. Enforcement of these laws against us could have a material adverse effect on our business, financial condition and results of operations.
We may become subject, directly or indirectly, to federal and state health care fraud and abuse laws and regulations and if we are unable to fully comply with such laws, the Company could face substantial penalties.
Although not affected at this time, our operations may in the future become directly or indirectly affected by various broad state and federal health care fraud and abuse laws, including the Federal Healthcare Programs’ Anti-Kickback Statute and the Stark law, which among other things, prohibits a physician from referring Medicare and Medicaid patients to an entity with which the physician has a financial relationship, subject to certain exceptions. If our future operations are found to be in violation of these laws, we or our officers may be subject to civil or criminal penalties, including large monetary penalties, damages, fines, imprisonment and exclusion from Medicare and Medicaid program participation. If enforcement action were to occur, our business and results of operations could be adversely affected.
We may be subject to federal and state false claims laws which impose substantial penalties.
Many of the physicians and patients whom we expect to use our services will file claims for reimbursement with government programs such as Medicare and Medicaid. As a result, we may be subject to the federal False Claims Act if we knowingly “cause” the filing of false claims. Violations may result in substantial civil penalties, including treble damages. The federal False Claims Act also contains “whistleblower” or “qui tam” provisions that allow private individuals to bring actions on behalf of the government alleging that the defendant has defrauded the government. In recent years, the number of suits brought in the medical industry by private individuals has increased dramatically. Various states have enacted laws modeled after the federal False Claims Act, including “qui tam” provisions, and some of these laws apply to claims filed with commercial insurers. We are unable to predict whether we could be subject to actions under the federal False Claims Act, or the impact of such actions. However, the costs of defending claims under the False Claims Act, as well as sanctions imposed under the False Claims Act, could adversely affect our results of operations.
Risks Related To Common Stock
The price of our Common Stock and Warrants may be subject to wide fluctuations.
A consistently active trading market for our Common Stock and Warrants does not exist and may not develop or be maintained. You may not be able to sell your shares quickly or at the current market price if trading in our stock is not active. You may lose all or a part of your investment. The market price of our Common Stock and Warrants may be highly volatile and subject to wide fluctuations in response to a variety of factors and risks, many of which are beyond our control. In addition to the risks noted elsewhere in this prospectus, some of the other factors affecting our stock price may include:
•variations in our operating results;
•announcements by us or our competitors of significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments;
•announcements by third parties of significant claims or proceedings against us;
•future sales of our Common Stock or Warrants;
•any delay in our regulatory filings for our product and any adverse development or perceived adverse development with respect to the applicable regulatory authority’s review of such filings, including without limitation the FDA’s issuance of a “refusal to file” letter or a request for additional information;
•adverse results or delays in clinical trials;
•our decision to initiate a clinical trial, not to initiate a clinical trial or to terminate an existing clinical trial;
•adverse regulatory decisions, including failure to receive regulatory approval of our product;
•changes in laws or regulations applicable to our products, including but not limited to clinical trial requirements for approvals;
•adverse developments concerning our manufacturers;
•our inability to obtain adequate product supply for any approved product or inability to do so at acceptable prices;
•our inability to establish collaborations if needed;
•additions or departures of key scientific or management personnel;
•introduction of new products or services offered by us or our competitors;
•announcements of significant acquisitions, strategic partnerships, joint ventures or capital commitments by us or our competitors;
•our ability to effectively manage our growth;
•the size and growth of our initial target markets;
•our ability to successfully treat additional types of indications or at different stages;
•actual or anticipated variations in annual and quarterly operating results;
•our cash position;
•our failure to meet the estimates and projections of the investment community or that we may otherwise provide to the public;
•publication of research reports about us or our industry, or positive or negative recommendations or withdrawal of research coverage by securities analysts;
•changes in the market valuations of similar companies;
•overall performance of the equity markets;
•sales of our Common Stock by us or our stockholders in the future;
•trading volume of our Common Stock;
•changes in accounting practices;
•ineffectiveness of our internal controls;
•disputes or other developments relating to proprietary rights, including patents, litigation matters and our ability to obtain patent protection for our or our licensee’s technologies;
•significant lawsuits, including patent or stockholder litigation;
•general political and economic conditions, including war and its unknown impact on our Serbia development team; and
•other events or factors, many of which are beyond our control.
We are an “emerging growth company,” and any decision on our part to comply with certain reduced disclosure requirements
We are an “emerging growth company” as defined in the JOBS Act, and, for as long as we continue to be an emerging growth company, we may choose to take advantage of exemptions from various reporting requirements applicable to other public companies including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, not being required to comply with any new requirements adopted by the Public Company Accounting Oversight Board (the “PCAOB”), requiring mandatory audit firm rotation or a supplement to the auditor’s report in which the auditor would be required to provide additional information about the audit and the financial statements of the issuer, not being required to comply with any new audit rules adopted by the PCAOB after April 5, 2012 unless the SEC determines otherwise, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could remain an emerging growth company until the earlier of: (i) the last day of the fiscal year in which we have total annual gross revenues of $1.07 billion or more; (ii) the last day of our fiscal year following the fifth anniversary of the date of our first sale of common equity securities pursuant to an effective registration statement; (iii) the date on which we have issued more than $1.0 billion in nonconvertible debt during the previous three years; or (iv) the date on which we are deemed to be a large accelerated filer. We cannot predict if investors will find our Units less attractive if we choose to rely on these exemptions. If some investors find our Units less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our Units and our stock price may be more volatile. Further, as a result of these scaled
regulatory requirements, our disclosure may be more limited than that of other public companies and you may not have the same protections afforded to shareholders of such companies.
Section 107 of the JOBS Act also provides that an emerging growth company can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended (the “Securities Act”), for complying with new or revised accounting standards. We have opted for taking advantage of the extended transition period for complying with new or revised accounting standards pursuant to Section 107(b) of the Jobs Act.
As a result of our becoming a public company, we will become subject to additional reporting and corporate governance requirements that will require additional management time, resources and expense.
As a public company, and particularly after we are no longer an emerging growth company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. The Sarbanes-Oxley Act, the Dodd-Frank Wall Street Reform and Consumer Protection Act, the listing requirements of the NASDAQ and other applicable securities rules and regulations impose various requirements on public companies, including the obligation to file with the SEC annual and quarterly information and other reports that are specified in the Securities Exchange Act of 1934, as amended (the “Exchange Act”), and to establish and maintain effective disclosure and financial controls and corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly.
We evaluate these rules and regulations, and cannot predict or estimate the amount of additional costs we may incur or the timing of such costs. These rules and regulations are often subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices.
Future sales and issuances of our Common Stock or rights to purchase Common Stock, including pursuant to our equity incentive plans and outstanding warrants could result in dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital may be needed in the future to continue our planned operations, including conducting clinical trials, commercialization efforts, expanded research and development activities and costs associated with operating a public company. To raise capital, we may sell Common Stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell Common Stock, convertible securities or other equity securities, investors may be materially diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights, preferences and privileges senior to the holders of our Common Stock. Our Certificate of Incorporation authorizes the issuance of 20,000,000 shares of Common Stock. As of March 23, 2022, there was 7,958,888 shares of Common Stock issued and outstanding. Initially, the aggregate number of shares of our Common Stock that may be issued pursuant to stock awards under our 2015 Plan is approximately 234,439 shares. Increases in the number of shares available for future grant or purchase may result in additional dilution, which could cause our stock price to decline. In addition, the issuance of shares of Common Stock upon conversion of the 2015 Convertible Notes, if converted at the option of the holder, will also have a dilutive effect on the percentage ownership held by holders of our Common Stock.
Nasdaq Capital Market, may delist our Common Stock if we fail to comply with ongoing listing standards.
Nasdaq Capital Market requires us to meet certain financial, public float, bid price and liquidity standards on an ongoing basis in order to continue the listing of our Common Stock and Warrants. If we fail to meet these continued listing requirements, our Common Stock or Warrants may be subject to delisting. If our Common Stock or Warrants are delisted and we are not able to list such Common Stock and Warrants on another national securities exchange, we expect our securities would be quoted on an over-the-counter market; However, if this were to occur, our stockholders could face significant material adverse consequences, including limited availability of market quotations for our Common Stock and Warrants and reduced liquidity for the trading of our securities. In addition, in the event of such delisting, we could experience a decreased ability to issue additional securities and obtain additional financing in the future.
If securities or industry analysts do not publish research or publish inaccurate or unfavorable research about our business, our stock price and trading volume could decline.
The trading market for our Common Stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. Securities and industry analysts do not currently, and may never, publish research
on our Company. If no securities or industry analysts commence coverage of our Company, the trading price for our stock would likely be negatively impacted. In the event securities or industry analysts initiate coverage, if one or more of the analysts who covers us downgrades our stock or publishes inaccurate or unfavorable research about our business, our stock price may decline. If one or more of these analysts ceases coverage of our Company or fails to publish reports on us regularly, demand for our stock could decrease, which might cause our stock price and trading volume to decline.
Our need for future financing may result in the issuance of additional securities which will cause investors to experience dilution.
Our cash requirements may vary from those now planned depending upon numerous factors, including the result of future research and development activities, our ability to estimate future expenses and acceptance of our products in the market.
There are no commitments for future financing of the commercial phase of our telehealth Product and other future products. Though we believe a successful ED Product introduction will be a significant value creation event for us, in the future, our securities may be offered to other investors at a price lower than the price per share paid by our investors, or upon terms which may be deemed more favorable than previously offered. In addition, the issuance of securities in any future financing using our securities may dilute an investor’s equity ownership. Moreover, we may issue derivative securities, including options and/or warrants, from time to time, to procure qualified personnel or for other business reasons. The issuance of any such derivative securities, which is at the discretion of our board of directors, may further dilute the equity ownership of our stockholders, including the investors in this offering. No assurance can be given as to our ability to procure additional financing, if required, and on terms deemed favorable to us. To the extent additional capital is required and cannot be raised successfully, we may then have to limit our then current operations and/or may have to curtail certain, if not all, of our business objectives and plans.
If our shares become subject to the penny stock rules, it would become more difficult to trade our shares.
The SEC has adopted regulations, which generally define “penny stock” to be an equity security that has a market price of less than $5.00 per share, subject to specific exemptions. The market price of our Common Stock is less than $5.00 per share and therefore may be a “penny stock.” Brokers and dealers effecting transactions in “penny stock” must disclose certain information concerning the transaction, obtain a written agreement from the purchaser and determine that the purchaser is reasonably suitable to purchase the securities. These rules may restrict the ability of brokers or dealers to sell our Common Stock and may affect your ability to sell shares of our Common Stock in the future.
Liability of directors for breach of duty is limited under Delaware law.
Our certificate of incorporation limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except for liability for any:
•breach of their duty of loyalty to us or our stockholders;
•act or omission not in good faith or that involves intentional misconduct or a knowing violation of law;
•unlawful payments of dividends or unlawful stock repurchases or redemptions as provided in Section 174 of the Delaware General Corporation Law; or
•transaction from which the directors derived an improper personal benefit.
These limitations of liability do not apply to liabilities arising under the federal or state securities laws and do not affect the availability of equitable remedies such as injunctive relief or rescission.
Our bylaws provide that we will indemnify for our directors and officers to the fullest extent permitted by law, and may indemnify employees and other agents. Our bylaws also provide that we are obligated to advance expenses incurred by a director or officer in advance of the final disposition of any action or proceeding.
We plan to enter into separate indemnification agreements with our directors and officers. These agreements, among other things, require us to indemnify our directors and officers for any and all expenses (including reasonable attorneys’ fees, retainers, court costs, transcript costs, fees of experts, witness fees, travel expenses, duplicating costs, printing and binding costs, telephone charges, postage, delivery service fees) judgments, fines and amounts paid in settlement actually and reasonably incurred by such directors or officers or on his or her behalf in connection with any action or proceeding arising out of their services as one of our directors or officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request provided that such person follows the procedures for determining entitlement to indemnification and advancement of expenses set forth in the indemnification agreement. We believe that these bylaw provisions and indemnification agreements are necessary to attract and retain qualified persons as directors and officers.
The limitation of liability and indemnification provisions in our certificate of incorporation and bylaws may discourage stockholders from bringing a lawsuit against directors for breach of their fiduciary duties. They may also reduce the likelihood of derivative litigation against directors and officers, even though an action, if successful, might provide a benefit to us and our stockholders. Our results of operations and financial condition may be harmed to the extent we pay the costs of settlement and damage awards against directors and officers pursuant to these indemnification provisions.
In so far as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling us, we have been informed that, in the opinion of the SEC, such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
At present, there is no pending litigation or proceeding involving any of our directors or officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
We do not anticipate paying any cash dividends on our Common Stock in the foreseeable future and, as such, capital appreciation, if any, of our Common Stock will be your sole source of gain for the foreseeable future.
We do not anticipate paying any cash dividends on our Common Stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. In addition, and any future loan arrangements we enter into may contain, terms prohibiting or limiting the amount of dividends that may be declared or paid on our Common Stock. As a result, capital appreciation, if any, of our Common Stock will be your sole source of gain for the foreseeable future.
Item 1B. Unresolved Staff Comments
None
Item 2. Properties
Our headquarters are located at 2118 Walsh Avenue, Suite 210, Santa Clara, CA 95050 which we lease pursuant to a monthly lease agreement entered into in May 2019. The terms of the lease are month to month and either party can terminate with one months notice.
We believe that the facilities described above are suitable and adequate for our present purposes and needs in the near future.
Item 3. Legal Proceedings.
There are no actions, suits, proceedings, inquiries or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of the executive officers of our company or any of our subsidiaries, threatened against or affecting our company, our Common Stock, any of our officers or directors in their capacities as such, in which an adverse decision could have a material adverse effect.
Item 4. Mine Safety Disclosures
Not applicable
Part II
Item 5. Market for Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
On November 11, 2021, our common stock and warrants issued in connection with our initial public offering (“IPO”) began trading on The NASDAQ Capital Market under the symbols “BEAT” and “BEATW,” respectively. Prior to our IPO, no public trades occurred in our common stock or warrants. The closing price of our common stock and warrants on the Nasdaq Capital Market on December 31, 2021 was $3.08 and $0.75, respectively.
Use of Proceeds
Use of Proceeds from the IPO
On November 10, 2021, we completed our IPO, in which we issued and sold 2,750,000 shares (the “Units”), with each Unit consisting of one share of Common Stock and one warrant (the “Warrants”) to purchase Common Stock at a combined public offering price of $6.00 per Unit. We received approximately $14,713,000 in net proceeds from the IPO after deducting the underwriting discount and commission and other IPO expenses payable by the Company of approximately $1,800,000. We intend to use the net proceeds from the offering to conduct operations, increase marketing efforts, and investments in the Company’s existing business initiatives and products, as well as general working capital.
Securities Authorized for Issuance Under Equity Compensation Plans
Information about our equity compensation plans is incorporated by reference herein to Item 12 of this Annual Report on Form 10-K.
Holders
As of March 23, 2022, there were approximately 77 holders of record of our common stock. As many of our shares of common stock are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of stockholders represented by these record holders.
Dividend Policy
We have not paid any cash dividends on our common stock to date. The payment of dividends in the future will be contingent upon our revenues and earnings, if any, capital requirements and general financial condition, and will be within the discretion of our then-existing board of directors.
Recent Sales of Unregistered Securities
On November 11, 2021 the Company issued 1,497,216 shares of Common Stock from the conversion of the 2015 Notes.
On November 15, 2021, in connection with the IPO, the Company issued 192,500 warrants (the “Underwriter Warrants”) to purchase Common Stock as compensation to the Underwriter, exercisable at a per share exercise price equal to $7.50 per share. The Underwriter Warrants will expire five years from the date of issuance and are subject to a 180-day lock-up period.
These issuances were made in reliance on an exemption from registration set forth in Section 4(a)(2) of the Securities Act,
as transactions by an issuer not involving a public offering.
Purchases of Equity Securities by the Issuer and Affiliated Purchasers
None
Item 6. SELECTED FINANCIAL DATA
We are a smaller reporting company as defined by Rule 12b-2 of the Securities Exchange Act of 1934 and are not required to provide the information under this item.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our financial statements and related notes appearing elsewhere in this Annual Report on Form 10-K. In addition to historical information, this discussion and analysis contains forward-looking statements that involve risks, uncertainties and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, including but not limited to, those set forth under “Risk Factors” and elsewhere in this Annual Report on Form 10-K. See Cautionary Statement Regarding Forward-Looking Statements.”
Overview
We are a medical technology company primarily focusing on telemedicine solutions that enable the detection and monitoring of cardiac disease outside a healthcare facility setting. Our aim is to deliver innovative, remote diagnostic and monitoring technologies that can be used for patients anywhere, with initial offerings for ambulatory and emergency department use. Our products require FDA clearance and have not been cleared for marketing (hereinafter “Product” or “Products”.) We believe our Products and services will benefit many stakeholders, including patients, healthcare providers, and healthcare payors. Our initial focus is providing diagnostic data to help physicians with care management of patients with cardiovascular disease. There are two major markets for our initial Products: remote patient monitoring and the hospital ED. We are developing our telehealth Product to address the rapidly growing field of remote patient monitoring. Our telehealth Product is comprised of a credit card sized ECG machine and a powerful cloud-based diagnostic software expert system. We believe that we are uniquely positioned to play a central role in remote monitoring of high-risk coronary artery disease patients, because the studies performed so far have shown that our ischemia detection system is highly accurate. Our powerful ischemia detection system is unlike other ambulatory cardiac monitors currently on the market which focus on arrhythmia detection. We are applying our platform technology to create a software tool for detecting heart attacks in the ED environment. This software tool is designed to enable emergency physicians to more accurately and quickly diagnose heart attacks than currently available. Market release of this Product will precede that of the telehealth Product.
To date, we have developed working prototypes for both our telehealth Product and our ED Product. The ED Product is currently undergoing additional engineering work that we believe will make it ready for FDA 510(k) clearance submission. Both Products have been validated in three medical studies, which were designed by Harvard Medical School faculty. Peer reviewed publications that describe the studies and results are in preparation. One peer reviewed medical publication submission is planned for the second quarter of 2022 and followed by another one later in 2022. These two publications will describe results of our two key studies: HeartBeam Ischemia Detection Study (“HIDES”) and B Score. In 2022 we plan to publish results of a study on performance of our ED Product in the real-life environment of an ED. In November 2021 we presented a technology abstract at the IEEE EMBS 2021 Conference. The abstract describes the technology foundation of our ED Product.
The custom software and hardware of our Products, we believe, are classified as Class II medical devices by the FDA running on an FDA approved Class I registered platform. Class II medical devices are those for which general controls alone are insufficient to provide reasonable assurance of safety and effectiveness and there is sufficient information to establish special controls. Special controls can include performance standards, post-market surveillance, patient histories and FDA guidance documents. Premarket review and clearance by the FDA for these devices is generally accomplished through the 510(k) or 510(k) de-novo premarket notification process
HeartBeam has three issued U.S. patents (U.S.10,433,744, U.S.10,117,592 and U.S.11,071,490), and six pending U.S applications. Three of the pending applications have published, all of the remaining three pending cases are unpublished. Outside of the U.S., HeartBeam has three pending EU utility patent applications (one of which is allowed), one pending CA utility patent application, one pending CN utility patent application, one pending JP utility patent application, one pending AU application, and two pending Patent Cooperation Treaty (“PCT”) applications.
As of December 31, 2021, we had 5 employees. We intend to hire or engage additional full-time professionals, employees, and / or consultants in alignment with our growth strategy. Although the market is highly competitive for attracting and retaining highly qualified professionals in our industry, we continue our endeavor to find such candidates for our Company. Our management team and additional personnel that we may hire in the future will be primarily responsible for executing and implementing growth opportunities, making tactical decisions related to our strategy and pursuing opportunities to invest in new technologies through strategic partnerships and acquisitions.
Significant Developments during 2021 and early 2022
Initial Public Offering
On November 10, 2021, our Registration Statement relating to our IPO was declared effective by the SEC. The IPO consisted of 2,750,000 Units, with each Unit consisting of one share of Common Stock, and one Firm Warrant to purchase Common Stock at a combined public offering price of $6.00 per Unit. The Common Stock and the Firm Warrants were immediately separable and issued separately but were purchased together in the IPO. The Firm Warrants will have a per share exercise price of $6.00 and are exercisable immediately. The Warrants will expire five years from the date of issuance.
Pursuant to the Underwriting Agreement dated November 10, 2021 between us and The Benchmark Company, LLC (the “Underwriter”) we granted the Underwriter a 30-day option to purchase up to an additional 412,500 shares of our Common Stock and/or Firm Warrants to cover over-allotments. On November 11, 2021, the Underwriter exercised its over-allotment option in respect of 412,500 Firm Warrants.
On November 10, 2021, as a result of the completion of the IPO and as required under the terms of the 2015 Notes, we converted the entirety of the outstanding principal of $5,084,000 and interest accrued of $1,204,404 to the 2015 Notes to 1,497,216 shares of common stock at the conversion price and issued shares to the 2015 Note holders, fully satisfying our obligations.
On November 15, 2021, we consummated the IPO and issued 192,500 warrants as compensation to the Underwriter, exercisable at a per share exercise price equal to $7.50 per share. The warrants will expire five years from the date of issuance and are subject to a 180-day lock-up period.
On November 15, 2021, we received approximately $14.7 million in net proceeds from the IPO after deducting the underwriting discount and commission and other estimated offering expenses payable by the Company.
During 2021, we raised an additional $1.7 million from the issuance of our 2015 Notes.
LIVMOR Partnership
On January 31, 2022, we entered into a partnership agreement (the “Partnership Agreement”) with LIVMOR to build a Company-branded version of the LIVMOR’s Halo+ FDA cleared turnkey solution for RPM to connect physicians and patients. The Company-branded version (“HeartBeam Platform”) of LIVMOR’s cloud-based remote monitoring portal will be an FDA registered Class 1 system and fully compliant with FDA standards for cybersecurity, software engineering and human factors and includes the prerequisite infrastructure for industry-leading solutions for telehealth. The Partnership Agreement also outlines rights and responsibilities for the customization of the HeartBeam Platform by LIVMOR with senior executives of both companies supervising the project. The Company’s Partnership Agreement with LIVMOR further support’s the Company’s project schedule for the FDA submission of its first product, an easy-to-use heart attack detection software solution, in an ED setting. In addition, the Partnership Agreement provides defined statement of work for development of the Company’s product, project management supervised by senior executives of the Company and LIVMOR, and regulatory support assistance for HeartBeam Platform by LIVMOR to obtain FDA clearance. Per the Partnership Agreement, the Company and LIVMOR have the right to enter into additional agreements as needed in order to further the Company’s development of its products.
Stock Purchase Agreement
On February 18, 2022, we entered into a stock purchase agreement (“Stock Purchase Agreement”) pursuant to which the Company agreed to issue and sell (“Private Placement”) to OpenSky Opportunities Fund Ltd. (“Purchaser”) an aggregate of 58,000 units (“Units”), with each Unit consisting of one share of common stock, par value $0.0001 per share (“Common Stock”) and one warrant (the “Warrants”) to purchase one share of Common Stock at a combined price of $6 per Unit. The Common Stock and the Warrants were immediately separable and issued separately but were purchased together in the
Private Placement. The Units, the Common Stock and the Warrants issued pursuant to the Stock Purchase Agreement shall be referred to as the “Securities”. We received $348,000 in proceeds from the Private Placement. The Warrants issued with the Private Placement will have a per share exercise price of $6.00 and are exercisable immediately subject to a 180-Day lock up. The Warrants will expire five years from the date of issuance. The Stock Purchase Agreement contains customary representations and warranties of the parties.
Technology Development Agreement
On March 7, 2022 we entered into a professional services agreement (the “Triple Ring Agreement”) with Triple Ring Technologies, Inc. (“Triple Ring”), a co-development company, to assist in the design and development of our telehealth complete solution 3D vector ECG collection device for remote heart attack or MI monitoring. The Triple Ring Agreement is a five-phase expedited device development project scheduled to be completed in the fourth quarter of 2022 for a 510k submission to the FDA.
Under the terms of the Triple Ring Agreement, the joint project will include our telehealth 3D vector ECG collection device builds for design verification and validation, device packaging, and a manufacturing technology transfer to a contract manufacturer to be named later.
The agreements with LIVMOR and Triple Ring include commitments in 2022 of approximately $3.0 million, of which approximately $1.4 million have been made to date. In addition, there will be license fees on the future commercial use of the platform.
Industry Trends and Outlook
The COVID-19 coronavirus pandemic has significantly impacted the delivery of healthcare. The transformation in daily life during the pandemic required healthcare systems to adjust how they delivered care and focus on patient convenience because of the social isolation measures. This change has resulted in innovations in virtual care and telehealth and opened new market opportunities for digital health platforms. During the pandemic the adoption rate of telehealth has increased dramatically, especially in cardiology, radiology, behavioral health, and online consultation.
The US telehealth market was $63B in 2020 and is projected to grow from $90B in 2021 to $636B in 2028 at a CAGR of 32% in the 2021-2028 period. The sudden growth in CAGR is attributable to the current market’s demand and growth, returning to pre-pandemic levels once the pandemic is over.
The change in clinical practice driven by the COVID-19 pandemic resulted in governments of many countries actively developing new policies and reimbursement guidelines to promote the use of digital health platforms. In the US, the Centers for Medicare & Medicaid Services (CMS) expanded reimbursement for telehealth. These market trends shifted care delivery from the traditional healthcare settings. Online audio and video consultations with physicians are becoming the new normal, resulting in reduced patient waiting times, easy access to specialists and a cost-effective solution for healthcare systems.
The active Government initiatives for telehealth solutions and the favorable policies to encourage telehealth solutions are set to propel market growth. Telehealth helps overcome the distance barrier for patients in rural locations and enables healthcare systems to provide virtual care platforms to serve patients with limited access to quality healthcare. The combination of market conditions, the rising prevalence of chronic conditions and the growing geriatric population point to the huge potential of telehealth. However, there are technological and infrastructure barriers that are key reasons inhibiting the expansion of the market, especially in developing countries.
We believe these favorable market conditions and our current progress on developing our first ED software product and our telehealth end-to-end solution for the patient and physician provide a positive commercial opportunity for the company. While there are technological and infrastructure barriers, especially in developing countries, our initial focus is on the US market and we don’t expect these issues to exist as we enter the US market.
Results of operations for the years ended December 31, 2021 and 2020
The following table summarizes our results of operations for the periods presented on our statement of operations data. The year over year comparison of results of operations is not necessarily indicative of results of operations for future periods.
| | | | | | | | | | | | | | | | | | | | | | | |
| Years ended December 31, |
| 2021 | | 2020 | | $ Change | | % Change |
| (In thousands, except percentages) |
| Operating expenses: | | | | | | | |
| General and administrative | $ | 2,030 | | | $ | 655 | | | $ | 1,375 | | | 210 | % |
| Research and development | 255 | | | 133 | | | 122 | | | 92 | % |
| Total operating expenses | 2,285 | | | 788 | | | 1,497 | | | 190 | % |
| | | | | | | |
| Loss from operations | (2,285) | | | (788) | | | (1,497) | | | 190 | % |
| | | | | | | |
| Interest expense | (2,165) | | | (280) | | | (1,885) | | | 673 | % |
| | | | | | | |
| Other income | 22 | | | — | | | 22 | | | 100 | % |
| | | | | | | |
| Income tax provision | — | | | — | | | — | | | — | % |
| | | | | | | |
| Net loss | $ | (4,428) | | | $ | (1,068) | | | $ | (3,360) | | | 315 | % |
Summary of Statements of Operations for the year ended December 31, 2021 compared with the year ended December 31, 2020:
General and administrative expenses (“G&A”) are largely related to personnel and professional services. G&A expense increased $1.4 million or 210% when compared to the same period in 2020. The primary increases were in employee headcount related expenses increasing $0.8 million, from $0.2 million to $1.0 million, related to the transition to market rate salaries and annual bonuses to certain executives during the fourth quarter of 2021, and business development, which included public company expenses, comprising of BOD fees, investor and public relations and SEC reporting.
Research and development expenses (“R&D”) are primarily from internally developed software and our credit-card sized collection device. R&D expense increased $0.1 million or 92% when compared with the same period in 2020. Our focus on R&D consisted largely of consultants associated with the development of our telehealth system and in 2021 the initial development work on the software-only ED Product.
Interest expense increased $1.9 million or 673%, primarily due to the accretion of the 2015 Notes 30% discount of approximately $1.9 million.
Other income of $22,000 during 2021 is due to the forgiveness of loans during the first quarter of 2021 issued under the Coronavirus Aid, Relief, and Economic Security Act (“CARES Act”).
Liquidity and Capital Resources
Our cash requirements are and will continue to be, dependent upon a variety of factors. We expect to continue devoting significant capital resources to R&D and go to market strategies.
As of December 31, 2021, we had approximately $13.2 million in cash, an increase of $13.18 million from $0.02 million as of December 31, 2020.
During the year ended December 31, 2021, we raised approximately $14.7 million in net proceeds from our IPO and an additional $1.7 million in 2015 Notes to support our on-going business plans.
Based on our current business plan assumptions and expected cash burn rate, we believe that our existing cash is sufficient to fund operations for the next twelve months.
Our cash is as follows (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | December 31, |
| | | 2021 | | 2020 |
| Cash | | $ | 13,192 | | | $ | 24 |
Cash flows for the year ended December 31, 2021 and 2020 (in thousands):
| | | | | | | | | | | | | | |
| | December 31 |
| | 2021 | | 2020 |
| Net cash used in operating activities | | (3,230) | | | (600) | |
| Net cash provided by financing activities | | $ | 16,398 | | | $ | 619 | |
Operating Activities:
Net cash used by our operating activities of $3.2 million during the year ended December 31, 2021, is primarily due to our net loss of $4.4 million less $2.3 million in non-cash expenses, offset by an increase of $1.1 million of net changes in operating assets and liabilities.
Net cash used by our operating activities of $0.6 million during the year ended December 31, 2020, is primarily due to our net loss of $1.1 million less $0.3 million in non-cash expense and $0.2 million from changes in operating assets and liabilities.
Financing Activities:
During the year ended December 31, 2021, net cash provided by financing activities of $16.4 million was primarily from our IPO totaling $14.7 million net and issuance of our 2015 Notes totaling $1.7 million.
During the year ended December 31, 2020, net cash provided by financing activities of $0.6 million was primarily from the issuance of our 2015 Notes.
We received a loan and grant through the CARES Act during the year ended December 31, 2020 for a total value of approximately $22,000. Both were forgiven in the first quarter of 2021.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements.
Critical Accounting Policies and Estimates
We believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating this “Management’s Discussion and Analysis of Financial Condition and Results of Operation.”
The discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or “U.S. GAAP.” The preparation of these financial statements in accordance with U.S. GAAP requires us to make estimates, assumptions and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an on-going basis, we evaluate our estimates, assumptions and judgments, including those related to revenue recognition, bad debts, inventories, warranties and income taxes. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities and our revenue recognition. Actual results may differ from these estimates under different assumptions or conditions and the impact of such differences may be material to our financial statements.
Critical accounting policies are those policies that, in management’s view, are most important in the portrayal of our financial condition and results of operations. The methods, estimates and judgments that we use in applying our accounting policies have a significant impact on the results that we report in our financial statements. These critical accounting policies require us to make difficult and subjective judgments, often as a result of the need to make estimates regarding matters that are inherently uncertain. Those critical accounting policies and estimates that require the most significant judgment are discussed further below. We consider our most critical accounting policies and estimates to be revenue recognition, gain on settlements, valuation of long lived assets, income taxes and valuation allowances against net deferred tax assets, derivative liabilities, stock-based compensation and accounting for business combinations-acquisition method accounting.
Stock-based compensation
The compensation cost for all stock-based awards is measured at the grant date, based on the fair value of the award, determined using a Black-Scholes option pricing model for stock options and fair value on the date of grant for non-vested restricted stock, and is recognized as an expense over the employee’s requisite service period (generally the vesting period of the equity award). Determining the fair value of stock-based awards at the grant date requires significant estimates and judgments, including estimating the market price volatility of our Common Stock, future employee stock option exercise behavior and requisite service periods. We account for forfeitures as they occur, stock-based compensation expense recognized in the financial statements is reduced by the awards forfeited.
A total of 1,105,938 stock options remain outstanding as of December 31, 2021, under the 2015 Plan. We expect to increase the number of employees and consultants to help execute our strategy in the medical device business and support our public company functions. Accordingly, we expect that future equity based awards will continue to be made under the 2015 Plan and future equity incentive plans to our directors, officers and other employees and consultants, as a result, to the extent relevant, we may incur non-cash, stock-based compensation expenses in future periods that may not be comparable to historical periods presented in our financial statements.
Recent Accounting Pronouncements
In December 2019, the FASB issued ASU No. 2019-12 - Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (“ASU 2019-12”). ASU 2019-12 is part of the FASB’s overall simplification initiative and seeks to simplify the accounting for income taxes by updating certain guidance and removing certain exceptions. The updated guidance is effective for fiscal years beginning after December 15, 2020 and interim periods within those fiscal years. We adopted this guidance on January 1, 2021. The impact to the financial statements following this guidance is deemed immaterial.
In October 2020, the FASB issued ASU 2020-10, Codification Improvements, which updates various codification topics by clarifying or improving disclosure requirements to align with the SEC’s regulations. We adopted ASU 2020-10 as of January 1, 2021. The impact to the financial statements following this guidance is deemed immaterial.
In June 2016, the FASB issued ASU 2016-13 “Financial Instruments-Credit Losses-Measurement of Credit Losses on Financial Instruments”. This guidance replaces the current incurred loss impairment methodology with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. The guidance applies to loans, accounts receivable, trade receivables and other financial assets measured at amortized cost, loan commitments, debt securities and beneficial interests in securitized financial assets, but the effect on the Company is projected to be limited to accounts receivable. In May 2019, the FASB issued ASU 2019-05 “Financial Instruments-Credit Losses (Topic 326)” which provides transition relief for companies adopting ASU 2016-13. This guidance amends ASU 2016-13 to allow companies to elect, upon adoption of ASU 2016-13, the fair value option on financial instruments that were previously recorded at amortized cost under certain circumstances. Companies are required to make this election on an instrument by instrument basis. The guidance will be effective for the fiscal year beginning January 1, 2023, including interim periods within that year.
In August 2020, the FASB issued ASU No. 2020-06 (“ASU 2020-06”) “Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity.” ASU 2020-06 will simplify the accounting for convertible instruments by reducing the number of accounting models for convertible debt instruments and convertible preferred stock. Limiting the accounting models will result in fewer embedded conversion features being separately recognized from the host contract as compared with current GAAP. Convertible instruments that continue to be subject to separation models are (1) those with embedded conversion features that are not clearly and closely related to the host
contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting and (2) convertible debt instruments issued with substantial premiums for which the premiums are recorded as paid-in capital. ASU 2020-06 also amends the guidance for the derivatives scope exception for contracts in an entity’s own equity to reduce form-over-substance-based accounting conclusions. ASU 2020-06 will be effective January 1, 2024, for the us. Early adoption is permitted, but no earlier than January 1, 2021, including interim periods within that year. We have not early adopted ASU 2020-06 and are currently evaluating the effects of the adoption, but currently believe the guidance will have no impact on our accounting.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk.
The Company is a smaller reporting company as defined by Rule 12b-2 of the Exchange Act and is not required to provide the information required under this item.
Item 8. Financial Statements and Supplementary Data
The financial statements and supplementary data of the Company required by this Item are described in Item 15 of this Annual Report on Form 10-K and are presented beginning on page F-1.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosures
None
Item 9A. Controls and Procedures
Evaluation of Disclosure Controls and Procedures
The Company has adopted and maintains disclosure controls and procedures that are designed to provide reasonable assurance that information required to be disclosed in the reports filed under the Exchange Act, such as this Annual Report on Form 10-K, is collected, recorded, processed, summarized and reported within the time periods specified in the rules of the SEC. The Company’s disclosure controls and procedures are also designed to ensure that such information is accumulated and communicated to management to allow timely decisions regarding required disclosure. As required under Exchange Act Rule 13a-15, the Company’s management, including the Chief Executive Officer (our principal executive officer) and our Chief Financial Officer (our principal financial officer), after evaluating the effectiveness of disclosure controls and procedures, identified material weaknesses in our internal control over financial reporting. The material weaknesses identified include (i) policies and procedures which are not yet adequately documented, (ii) lack of proper approval processes and review processes and documentation for such reviews, (iii) insufficient GAAP experience regarding complex transactions and reporting, and (iv) insufficient number of staff to maintain optimal segregation of duties and levels of oversight. The Company intends to retain additional individuals and resources to remedy the ineffective controls.
Management’s Annual Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act. Our internal control over financial reporting includes those policies and procedures that:
•pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets;
•provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
•provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
This Annual Report on Form 10-K does not include an audit or attestation report from our registered public accounting firm regarding our internal control over financial reporting. Our management’s report was not subject to audit or attestation by our registered public accounting firm since we are not an accelerated filer or a large accelerated filer as defined in Rule 12b-2 under the Securities Exchange Act of 1934.
Changes in Internal Control
There were no changes in our internal control over financial reporting (as such term is defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) identified in connection with the evaluation required by Rule 13a-15(d) and 15d-15(d) of the Exchange Act that occurred during the fourth quarter of the year ended December 31, 2021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
None
Item 9C. Disclosure Regarding Foreign Jurisdiction that Prevent Inspections.
Not applicable
Part III
Item 10. Directors, Executive Officers and Corporate Governance
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2021. Information relating to this item will be included in an amendment to this Annual Report on Form 10-K if the Company’s definitive proxy statement is not filed within such time.
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934, requires our directors, executive officers and beneficial owners of more than 10% of our common stock to file with the SEC certain reports regarding their ownership of common stock or any changes in such ownership. Based on our own review, we believe that there were no late filings during 2021 other than: Mr. Richard Ferrari one late form 3 filing in which 3 transactions were reported late, Mrs. Marga Ortigas-Wedekind had one late form 3 filing in which 3 transactions were reported late, Mr. Willem Elfrink had one late form 3 in which 7 transactions were reported late, Mr. Branislav Vajdic had one late form 3 in which 5 transactions were reported late and one form 4 filing in which one transaction was reported late, Mr. Richard Brounstein had one late form 3 filing in which 5 transactions were reported late and Mr. Jon Hunt had one late form 3 filing in which 1 transaction was reported late and one late form 4 filing in which one transaction was reported late.
Item 11. Executive Compensation
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2021. Information relating to this item will be included in an amendment to this Annual Report on Form 10-K if the Company’s definitive proxy statement is not filed within such time.
Item 12. Security Ownership of Certain Beneficial Owner and Management and Related Stockholder Matters
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2021. Information relating to this item will be included in an amendment to this Annual Report on Form 10-K if the Company’s definitive proxy statement is not filed within such time.
Item 13. Certain Relationships and Related Transactions, and Director Independence
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2021. Information relating to this item will be included in an amendment to this Annual Report on Form 10-K if the Company’s definitive proxy statement is not filed within such time.
Item 14. Principal Accounting Fees and Services
The information required under this item is incorporated herein by reference to our definitive proxy statement pursuant to Regulation 14A, which proxy statement will be filed with the SEC not later than 120 days after the close of our fiscal year ended December 31, 2021. Information relating to this item will be included in an amendment to this Annual Report on Form 10-K if the Company’s definitive proxy statement is not filed within such time.
Part IV
Item 15. Exhibits and Financial Statement Schedules
| | | | | | | | |
Exhibit
Number | | Exhibit Description |
| 3.1 | | |
| 3.2 | | |
| 3.3 | | |
| 4.1 | | |
| 4.2 | | |
| 4.3 | | |
| 4.4 | | |
| 4.5 | | |
| 4.6 | | |
| 4.7 | | |
| 4.8 | | |
| 4.9 | | |
| 4.10 | | |
| 4.11 | | |
| 4.12 | | |
| 4.13 | | |
| 4.14 | | |
| 10.1 | | |
| 10.2 | | |
| 10.3 | | |
| 10.4 | | |
| 10.5 | | |
| 10.6 | | |
| 10.7 | | |
| 23.1 | | |
| 31.1 | | |
| | | | | | | | |
| 31.2 | | |
| 32.1 | | |
| 32.2 | | |
| 101.INS | | XBRL Instance Document*+ |
| 101.SCH | | XBRL Taxonomy Extension Schema Document*+ |
| 101.CAL | | XBRL Taxonomy Extension Calculation Linkbase Document*+ |
| 101.DEF | | XBRL Taxonomy Extension Definition Linkbase Document*+ |
| 101.LAB | | XBRL Taxonomy Extension Label Linkbase Document*+ |
| 101.PRE | | XBRL Taxonomy Extension Presentation Linkbase Document*+ |
| 104 | | Cover Page Interactive Data File - The cover page iXBRL tags are embedded within the inline XBRL document. |
| | |
| * | | Filed herewith. |
| † | | Management or compensatory plan or arrangement. |
| # | | This certification is being furnished and shall not be deemed “filed” with the SEC for purposes of Section 18 of the Exchange Act, or otherwise subject to the liability of that section, and shall not be deemed to be incorporated by reference into any filing under the Securities Act or the Exchange Act, except to the extent that the registrant specifically incorporates it by reference. |
| + | | Pursuant to Rule 406T of Regulation S-T, the Interactive Data Files in Exhibit 101 hereto are deemed not filed or part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, are deemed not filed for purposes of Section 18 of the Securities and Exchange Act of 1934, as amended, and otherwise are not subject to liability under those sections. |
Item 16. Form 10-K Summary
Not Applicable
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | | | | | | | |
| HEARTBEAM, Inc. |
| | |
| By: | /s/ Branislav Vajdic |
| Name: | Branislav Vajdic |
| Title: | Chief Executive Officer |
| | (Principal Executive Officer) |
| | |
| By: | /s/ Richard Brounstein |
| Name: | Richard Brounstein |
| Title: | Chief Financial Officer |
| Dated: March 24, 2022 | | (Principal Financial and Accounting Officer) |
POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below hereby constitutes and appoints Branislav Vajdic and Richard Brounstein and each of them, jointly and severally, his attorneys-in-fact, each with the power of substitution, for him in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K and to file the same, with exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that each of said attorneys-in-fact, or his substitute or substitutes, may do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated:
| | | | | | | | | | | | | | |
| Name | | Title | | Date |
| | | | |
| /s/ Richard Ferrari | | Executive Chairman | | March 24, 2022 |
| Richard Ferrari | | | | |
| /s/ George deUrioste | | Director | | March 24, 2022 |
| George deUrioste | | | | |
| /s/ Marga Ortigas-Wedekind | | Director | | March 24, 2022 |
| Marga Ortigas-Wedekind | | | | |
| /s/ Willem Elfrink | | Director | | March 24, 2022 |
| Willem Elfrink | | | | |
| /s/ Branislav Vajdic | | President & Chief Executive Officer | | March 24, 2022 |
| Branislav Vajdic | | (Principal Executive Officer) | | |
| /s/ Richard Brounstein | | Chief Financial Officer & Treasurer | | March 24, 2022 |
| Richard Brounstein | | (Principal Financial and Accounting Officer) | | |
Contents
| | | | | |
| Page |
Report of Independent Registered Public Accounting Firm (PCAOB ID No: 711) | |
| |
| Balance Sheets as of December 31, 2021 and 2020 | |
| |
| Statements of Operations for the years ended December 31, 2021 and 2020 | |
| |
| Statements of Changes in Stockholders’ Equity (Deficit) for the years ended December 31, 2021 and 2020 | |
| |
| Statements of Cash Flows for the years ended December 31, 2021 and 2020 | |
| |
| Notes to Financial Statements | |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of HeartBeam, Inc.
Opinion on the Financial Statements
We have audited the accompanying balance sheets of HeartBeam, Inc. (the Company) as of December 31, 2021 and 2020, and the related statements of operations, stockholders’ equity (deficit), and cash flows for each of the years in the two-year period ended December 31, 2021, and the related notes (collectively referred to as the financial statements). In our opinion, the financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the financial statements. We believe that our audits provide a reasonable basis for our opinion.
| | | | | |
/s/ Friedman LLP
We have served as the Company’s auditor since 2020. |
| |
| East Hanover, New Jersey |
| March 24, 2022 | |
| |
HEARTBEAM, INC.
Balance Sheets
(In thousands, except share data)
| | | | | | | | | | | |
| December 31, |
| 2021 | | 2020 |
| Assets | | | |
| Current Assets: | | | |
Cash | $ | 13,192 | | | $ | 24 | |
Prepaid expenses and other assets | 806 | | | 27 | |
Total Assets | $ | 13,998 | | | $ | 51 | |
| | | |
| Liabilities and Stockholders’ Equity (Deficit) | | | |
| Current Liabilities: | | | |
Accounts payable and accrued expenses (includes related party $1 and $15, respectively) | 588 | | | 489 | |
Convertible notes, net | — | | | 4,295 | |
Other - current liabilities | — | | | 52 | |
Total Liabilities | 588 | | | 4,836 | |
|
| | |
| Commitments and contingencies (Note 8) | | | |
| | | |
| Stockholders’ Equity (Deficit) | | | |
Common stock - $0.0001 par value; 20,000,000 shares authorized; 7,809,912 and 3,527,850 shares issued and outstanding at December 31, 2021 and 2020 | 1 | | | — | |
Additional paid in capital | 22,633 | | | 11 | |
Accumulated deficit | (9,224) | | | (4,796) | |
Total Stockholders’ Equity (Deficit) | $ | 13,410 | | | $ | (4,785) | |
| | | |
Total Liabilities and Stockholders’ Equity (Deficit) | $ | 13,998 | | | $ | 51 | |
See accompanying notes to the financial statements
HEARTBEAM, INC.
Statements of Operations
(In thousands, except share and per share data)
| | | | | | | | | | | | | | | | | |
| | | December 31, |
| | | | | 2021 | | 2020 |
| Operating Expenses: | | | | | | | |
General and administrative | | | | | $ | 2,030 | | | $ | 655 | |
Research and development | | | | | 255 | | | 133 | |
Total operating expenses | | | | | 2,285 | | | 788 | |
| | | | | | | |
| Loss from operations | | | | | (2,285) | | | (788) | |
| | | | | | | |
| Other Income (Expense) | | | | | | | |
| Interest expense | | | | | (2,165) | | | (280) | |
| Other Income | | | | | 22 | | | — | |
| Total other income (expense) | | | | | (2,143) | | | (280) | |
| | | | | | | |
| Loss before provision for income taxes | | | | | (4,428) | | | (1,068) | |
| | | | | | | |
| Income tax provision | | | | | — | | | — | |
| | | | | | | |
| Net Loss | | | | | $ | (4,428) | | | $ | (1,068) | |
| | | | | | | |
| Net loss per share, basic and diluted | | | | | $ | (1.03) | | | $ | (0.29) | |
| | | | | | | |
| Weighted average common shares outstanding, basic and diluted | | | | | 4,284,714 | | | 3,645,944 | |
See accompanying notes to the financial statements
HEARTBEAM, INC.
Statement of Changes in Stockholders’ Equity (Deficit)
(In thousands, except share data)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-in
Capital | | Accumulated
Deficit | | Total
Stockholders'
Equity (Deficit) |
| Shares | | Amount | | | |
| Balance – December 31, 2019 | 3,482,850 | | | $ | — | | | $ | 1 | | | $ | (3,728) | | | $ | (3,727) | |
| Stock based compensation, expense | — | | | — | | | 10 | | | — | | | 10 | |
| Common stock issued upon vesting and exercise of stock options | 45,000 | | | — | | | — | | | — | | | — | |
| Net loss | — | | | — | | | — | | | (1,068) | | | (1,068) | |
| Balance – December 31, 2020 | 3,527,850 | | | $ | — | | | $ | 11 | | | $ | (4,796) | | | $ | (4,785) | |
| | | | | | | | | |
| Stock based compensation, expense | — | | | — | | | 192 | | | — | | | 192 | |
| Common stock issued upon vesting and exercise of stock options | 34,846 | | | — | | | — | | | — | | | — | |
| Sale of Common Stock & Warrants, net of fees | 2,750,000 | | | 1 | | | 14,256 | | | — | | | 14,257 | |
| Common stock issuance upon conversion of 2015 Notes | 1,497,216 | | | — | | | 8,174 | | | — | | | 8,174 | |
| Net loss | — | | | — | | | — | | | (4,428) | | | (4,428) | |
| Balance – December 31, 2021 | 7,809,912 | | | $ | 1 | | | $ | 22,633 | | | $ | (9,224) | | | $ | 13,410 | |
See accompanying notes to the financial statements
HEARTBEAM, INC.
Statements of Cash Flows
(In thousands)
| | | | | | | | | | | |
| December 31, |
| 2021 | | 2020 |
| Cash Flows From Operating Activities | | | |
Net loss | $ | (4,428) | | | $ | (1,068) | |
Adjustments to reconcile net loss to net cash used in operating activities |
| | |
Accretion expense, convertible notes | 1,886 | | | — | |
Non-cash interest expense | 278 | | | 248 | |
Stock-based compensation expense | 192 | | | 10 | |
Amortization of debt issuance cost | — | | | 28 | |
PPP loan forgiveness | (22) | | | — | |
Changes in operating assets and liabilities: |
| | |
Prepaid expenses and other current assets | (779) | | | (25) | |
Accounts payable, accrued expenses and other current liabilities | (357) | | | 207 | |
Net cash used in operating activities | (3,230) | | | (600) | |
|
| | |
| Cash Flows From Financing Activities | | | |
| Proceeds from sale of equity in IPO, net | 14,713 | | | — | |
| Proceeds from issuance of convertible notes | 1,715 | | | 617 | |
| Proceeds from PPP & EIDL Loans | — | | | 22 | |
| Repayment and interest paid on short-term loans | (30) | | | (20) | |
| Net cash provided by financing activities | 16,398 | | | 619 | |
|
| | |
Net increase in cash | 13,168 | | | 19 | |
| Cash – Beginning of the year | 24 | | | 5 | |
| | | |
| Cash – End of the year | $ | 13,192 | | | $ | 24 | |
| | | |
| Supplemental Disclosures of Cash Flow Information: | | | |
Taxes paid | $ | — | | | $ | — | |
Interest paid | — | | | 4 | |
|
| | |
Supplemental Disclosures of Non-cash Flow Information: | | | |
| Conversion of debt to equity | 6,288 | | | — | |
Debt discount | 1,886 | | | — | |
| Common stock and awards accrued but not issued | 456 | | | — | |
| Non-cash - accounts payable converted to short term debt | — | | | 30 | |
Conversion of short-term notes to convertible notes | $ | — | | | $ | 22 | |
See accompanying notes to the financial statements
HEARTBEAM, INC.
NOTES TO FINANCIAL STATEMENTS
NOTE 1 – ORGANIZATION AND OPERATIONS
HeartBeam, Inc. (“HeartBeam” or the “Company”) is a development-stage company specializing in cardiovascular diagnostic technology. The Company was incorporated in 2015 as a Delaware corporation. The Company’s operations are based in Santa Clara, California and it operates in one segment.
HeartBeam’s initial focus is on timely diagnosis of a heart attack. The Company’s technology provides physicians with complete cardiac diagnostic information for a patient that is outside of a medical institution. The Electrocardiogram (“ECG”) collection device is the size of a credit card. The device sends ECG signals to the patient's smartphone and on to a cloud-based software expert system. Results of the cloud-based analysis are presented to a qualified health care professional for immediate action including, if necessary, a telehealth visit. The Company has validated this novel technology in three clinical studies and is preparing to seek U.S. Food and Drug Administration (“FDA”) clearance of its initial product during the second quarter of 2022. Clearance for the ED product is expected Q3 2022, submission of the telehealth product is during the latter part of 2022 with clearance expected early 2023.
On September 27, 2021, the Company filed a certificate of amendment to its amended and restated certificate of incorporation with the Secretary of State of the State of Delaware to effect a 1-for-2.75 reverse stock split of its outstanding shares of common stock. As a result of the reverse stock split, every 2.75 shares of the Company’s outstanding pre-reverse split common stock were combined and reclassified into one share of common stock. Unless otherwise noted, all share and per share data included in these financial statements retroactively reflect the 1-for-2.75 reverse stock split.
NOTE 2 – LIQUIDITY
The Company is subject to a number of risks similar to those of early stage companies, including dependence on key individuals and products, the difficulties inherent in the development of a commercial market, the potential need to obtain additional capital, competition from larger companies, other technology companies and other technologies.
The Company has incurred losses each year since inception and has experienced negative cash flows from operations in each year since inception. As of December 31, 2021 and December 31, 2020, the Company had an accumulated deficit of approximately $9,224,000 and $4,796,000, respectively. In November 2021, the Company raised approximately $14,713,000 from the completion of the initial public offering (the “IPO”) (see Note 5) and in February 2022, the Company raised an additional $348,000 from the issuance of stock through a stock purchase agreement (see Note 10). Based on its current business plan assumptions and expected cash burn rate, the Company believes that the existing cash is sufficient to fund operations for the next twelve months following the issuance of these financial statements.
The Company’s continued operations will depend on its ability to raise additional capital through various potential sources, such as equity and/or debt financings, or strategic relationships. Management can provide no assurance that such financing or strategic relationships will be available on acceptable terms, or at all, which would likely have a material adverse effect on the Company and its financial statements.
On January 30, 2020, the World Health Organization (“WHO”) announced a global health emergency, a result of the new strain of coronavirus (the “COVID-19 outbreak”) and the risks to the international community as the virus spreads globally beyond its point of origin. In March 2020, the WHO classified the COVID-19 outbreak as a pandemic, based on the rapid increase in exposure globally. There are many variables and uncertainties regarding the COVID -19 pandemic, including the emergence, contagiousness and threat of new and different strains of the virus and their severity; the effectiveness of treatments or vaccines against the virus or its new strains; the extent of travel restrictions, business closures and other measures that are or may be imposed in affected areas or countries by governmental authorities; disruptions in the supply chain; an increasingly competitive labor market due to a sustained labor shortage or increased turnover caused by the COVID -19 pandemic; increased logistics costs; additional costs due to remote working arrangements, adherence to social distancing guidelines and other COVID-19 related challenges.
NOTE 3 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
BASIS OF PRESENTATION
The accompanying financial statements have been prepared in conformity with US Generally Accepted Accounting Principles ("US GAAP") and have been prepared on a basis which assumes that the Company will continue as a going concern, which contemplates the realization of assets and the satisfaction of liabilities and commitments in the normal course of business.
USE OF ESTIMATES
The preparation of financial statements in conformity with US GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of expenses during the reporting period. Actual results could differ from those estimates. Due to the inherent uncertainty involved in making estimates, actual results reported in future periods may be based on amounts that differ from those estimates.
CASH AND CASH EQUIVALENTS
The Company considers all highly liquid investments with original maturities of three months or less to be cash equivalents. As of December 31, 2021 and 2020 there were no cash equivalents. The Company maintains cash balances in accounts which exceed the federally insured limits during the year ended December 31, 2021, management does not believe this results in any significant credit risk. As of December 31, 2020 there were no deposits at banks in excess of FDIC insured limits.
RESEARCH AND DEVELOPMENT EXPENSE
The Company expenses the cost of research and development as incurred. Research and development expenses consist primarily of professional services costs associated with the development of cardiovascular technologies and products.
FAIR VALUE OF FINANCIAL INSTRUMENTS
The Company’s financial instruments consist primarily of cash, accounts payable, accrued liabilities and debt instruments.
Fair value is defined as the exchange price that would be received for an asset or paid to transfer a liability in the principal or most advantageous market for the asset transaction between market participants on the measurement date. Where available, fair value is based on observable market prices or is derived from such prices. The Company uses the market approach valuation technique to value its investments. The market approach uses prices and other pertinent information generated from market transactions involving identical or comparable assets or liabilities. The types of factors that the Company may consider in fair value pricing the investments include available current market data, including relevant and applicable market quotes.
Financial assets and liabilities carried at fair value are classified and disclosed in one of the following three categories:
•Level 1 - Observable inputs such as quoted prices in active markets.
•Level 2 - Inputs, other than the quoted prices in active markets, that are observable either directly or indirectly.
•Level 3 - Unobservable inputs in which there is little or no market data, which require the reporting entity to develop its own assumptions.
In certain cases, the inputs used to measure fair value may fall into different levels of the fair value hierarchy. In such cases, the assignment of an asset or liability within the fair value hierarchy is based on the lowest level of input that is significant to the fair value measurement. The Company’s assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment and considers factors specific to the asset or liability.
ACCOUNTING FOR WARRANTS
The Company classifies as equity any contracts that (i) require physical settlement or net-share settlement or (ii) gives the Company a choice of net-cash settlement or settlement in its own shares (physical settlement or net-share settlement). The Company classifies as assets or liabilities any contracts that (i) require net-cash settlement (including a requirement to net-cash settle the contract if an event occurs and if that event is outside the control of the Company) or (ii) gives the counterparty a choice of net-cash settlement or settlement in shares (physical settlement or net-share settlement).
The Company accounts for its currently issued warrant instruments in conjunction with the Company’s common stock in permanent equity. These warrants are indexed to the Company’s stock and meet the requirements of equity classification as prescribed under ASC 815. Warrants classified as equity are initially measured at fair value, and subsequent changes in fair value are not recognized so long as the warrants continue to be classified as equity.
STOCK-BASED COMPENSATION
The Company periodically issues stock options and restricted stock awards to employees and non-employees for services. The Company has adopted ASU 2018-07 which expands the scope of Topic 718 to include share-based payment transactions for acquiring goods and services from non-employees. The Company accounts for such grants issued and vesting to employees and non-employees based on ASC 718, whereby the value of the award is measured on the date of grant and recognized as compensation expense over the vesting period.
The Company grants certain option holders the right to early exercise, as of December 31, 2021, 24,623 have been early exercised and remain unvested. These early exercised grants are not included in either shares outstanding or weighted average shares outstanding until vested.
The fair value of stock options on the date of grant is calculated using the Black-Scholes option pricing model, based on key assumptions such as the fair value of common stock, expected volatility and expected term. These estimates require the input of subjective assumptions, including (i) the expected stock price volatility, (ii) the calculation of the expected term of the award, (iii) the risk-free interest rate and (iv) expected dividends. These assumptions are primarily based on third-party valuations, historical data, peer company data and the judgment of management regarding future trends and other factors. The Company has estimated the expected term of its employee stock options using the “simplified” method, whereby, the expected term equals the arithmetic average of the vesting term and the original contractual term of the option due to its lack of sufficient historical data. The risk-free interest rates for periods within the expected term of the option are based on the US Treasury securities with a maturity date commensurate with the expected term of the associated award. The Company has never paid and does not expect to pay dividends in the foreseeable future. The Company accounts for forfeitures when they occur. Stock-based compensation expense recognized in the financial statements is reduced by the actual awards forfeited.
Compensation cost for restricted stock awards issued to employees and non-employees is measured using the grant date fair value of the award, and expense is recognized over the service period, adjusted to reflect actual forfeitures.
INCOME TAXES
The Company accounts for income taxes using the asset and liability method. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax basis and tax carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
A valuation allowance is established to reduce net deferred tax assets to the amount expected to be realized The Company recognizes the effect of income tax positions only if those positions are more likely than not of being sustained. Recognized income tax positions are measured at the largest amount that is greater than 50% likely of being recognized. Changes in recognition and measurement are reflected in the period in which the change in judgment occurs. Interest and penalties related to unrecognized tax benefits are included in income tax expense.
NET LOSS PER COMMON SHARE
Basic net loss per share excludes the effect of dilution and is computed by dividing the net loss attributable to common stockholders by the weighted-average number of shares of common stock outstanding.
Diluted net loss per share is computed by giving effect to all potential shares of common stock, including stock options and warrants to the extent dilutive. Basic net loss per share was the same as diluted net loss per share for the year ended December 31, 2021 and 2020 as the inclusion of all potential common shares outstanding would have an anti-dilutive effect.
As of December 31, 2021 and 2020, the penny warrants issued during 2019 have been excluded from the net loss per common share calculation following ASC 260-1-25-12A (Treatment of Contingently Issuable Shares in Weighted-Average Shares Outstanding), there are circumstances under which these shares would not be issued and therefore not exercisable, (see NOTE 5).
In accordance with ASC 260-10-45-13, exercisable penny options were included in the calculation of weighted average basic and diluted earnings per share.
The following is a summary of awards outstanding as of December 31, 2021 and 2020, which are not included in the computation of basic and diluted weighted average shares:
| | | | | | | | | | | | | | | |
| | | Year ended December 31, |
| | | | 2021 | | 2020 |
| Stock options (excluding exercisable penny stock options) | | | | | 936,996 | | | 318,034 | |
| Warrants | | | | | 3,777,549 | | | 422,549 | |
| Total | | | | | 4,714,545 | | | 740,583 | |
RECENTLY ISSUED ACCOUNTING STANDARDS
Adopted:
In December 2019, the FASB issued ASU No. 2019-12 - Income Taxes (Topic 740): Simplifying the Accounting for Income Taxes (“ASU 2019-12”). ASU 2019-12 is part of the FASB’s overall simplification initiative and seeks to simplify the accounting for income taxes by updating certain guidance and removing certain exceptions. The updated guidance is effective for fiscal years beginning after December 15, 2020 and interim periods within those fiscal years. The Company adopted this guidance on January 1, 2021. The impact to the financial statements following this guidance is deemed immaterial.
In October 2020, the FASB issued ASU 2020-10, Codification Improvements, which updates various codification topics by clarifying or improving disclosure requirements to align with the SEC’s regulations. The Company adopted ASU 2020-10 as of January 1, 2021. The impact to the financial statements following this guidance is deemed immaterial.
Not Yet Adopted as of December 31, 2021:
In June 2016, the FASB issued ASU 2016-13 “Financial Instruments-Credit Losses-Measurement of Credit Losses on Financial Instruments”. This guidance replaces the current incurred loss impairment methodology with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. The guidance applies to loans, accounts receivable, trade receivables and other financial assets measured at amortized cost, loan commitments, debt securities and beneficial interests in securitized financial assets, but the effect on the Company is projected to be limited to accounts receivable. In May 2019, the FASB issued ASU 2019-05 “Financial Instruments-Credit Losses (Topic 326)” which provides transition relief for companies adopting ASU 2016-13. This guidance amends ASU 2016-13 to allow companies to elect, upon adoption of ASU 2016-13, the fair value option on financial instruments that were previously recorded at amortized cost under certain circumstances. Companies are required to make this election on an instrument by instrument basis. The guidance will be effective for the fiscal year beginning January 1, 2023, including interim periods within that year.
In August 2020, the FASB issued ASU No. 2020-06 (“ASU 2020-06”) “Debt—Debt with Conversion and Other Options (Subtopic 470-20) and Derivatives and Hedging—Contracts in Entity’s Own Equity (Subtopic 815-40): Accounting for Convertible Instruments and Contracts in an Entity’s Own Equity.” ASU 2020-06 will simplify the accounting for convertible instruments by reducing the number of accounting models for convertible debt instruments and convertible preferred stock. Limiting the accounting models will result in fewer embedded conversion features being separately recognized from the host contract as compared with current GAAP. Convertible instruments that continue to be subject to separation models are (1) those with embedded conversion features that are not clearly and closely related to the host contract, that meet the definition of a derivative, and that do not qualify for a scope exception from derivative accounting and (2) convertible debt instruments issued with substantial premiums for which the premiums are recorded as paid-in capital. ASU 2020-06 also amends the guidance for the derivatives scope exception for contracts in an entity’s own equity to reduce form-over-substance-based accounting conclusions. ASU 2020-06 will be effective January 1, 2024, for the Company. Early adoption is permitted, but no earlier than January 1, 2021, including interim periods within that year. Management has not early adopted ASU 2020-06 and is currently evaluating the effects of the adoption, but currently believes the guidance will not have a significant impact on the Company’s accounting.
NOTE 4 – DEBT
CONVERTIBLE NOTES
On August 21, 2015, the Board of Directors approved the 2015 Note Subscription Agreement (the “2015 Notes”) authorizing financing through the sale and issuance of 2015 convertible promissory notes (the “Financing”) for an aggregate amount not to exceed $1,000,000, with a maturity date of August, 25, 2017, which was derived from the issuance of the first 2015 Note. The Company entered into a series of amendments over the years, of which the most recent during 2021 include the sixth amendment on March 22, 2021, expanding the definition of a Qualified Financing of at least $2,000,000 as defined in the 2015 Notes to include either preferred stock or common stock, followed by the seventh and final amendment on October 7, 2021, increasing the aggregate amount for issuance to $5,500,000. All amendments were updated in accordance with the 2015 Note Subscription Agreements and approved by the Board of Directors. The Company has accounted for the last amendment to the 2015 Notes in accordance with ASC 470-50-40-6, (modifications and exchanges), under modification accounting and there was no impact to the financial statements as a result of the amendment to the 2015 Notes.
The sale and purchase of the 2015 Notes take place at closing on the date of the agreements. At closing, the Company will deliver to the investor the 2015 Note to be purchased by such investor, against receipt by the Company of the corresponding purchase price. The 2015 Notes will be registered in each investor’s name in the Company’s records. The 2015 Notes accrue interest payable at the rate of eight percent (8%) and the conversion price is equal to seventy percent (70%) of the per share price at which shares of preferred stock or common stock is to be sold.
On November 10, 2021, as a result of the completion of the IPO (see Note 5) and as required under the terms of the 2015 Notes, the Company converted the entirety of the outstanding principal of $5,084,000 and interest accrued of $1,204,404 to 1,497,216 shares of common stock at the Conversion Price of $4.20 per share and issued the shares to the 2015 Note holders, fully satisfying the Company’s obligations.
The Company assessed the probability of a Qualified Financing occurring before maturity of the 2015 Notes to be greater than 50% (more likely than not). In accordance with the guidance ASC 480, the Company recorded the amount of the 2015 Notes’ 30% conversion discount of the sum of principal and accrued interest to the earliest of conversion date (if known) or maturity. As of December 31, 2021, the Company recorded approximately $1,886,000 as debt discount. As all of the debt converted at the IPO, the Company fully accreted the debt discount as interest expense as of December 31, 2021.
As of December 31, 2020, the Company had $3,369,000 in 2015 Notes, and $926,000 in accrued interest.
In 2019, the Company incurred financing fees of approximately $64,000. Under guidance ASC 835-30-35, these costs have been amortized and recognized as interest expense over the life of the 2015 Notes. During the year ended December 31, 2020, the Company amortized approximately $28,000 of the remaining balance as interest expense. There was no such expense during the year ended December 31, 2021.
SHORT TERM LOANS
During 2020, the Company settled its short term notes liability of $42,000. As settlement, the Company converted $22,000 into the 2015 Notes and the remaining $20,000, which included the amended $10,000 promissory note, a $6,000 advance and $4,000 in accrued interest, was repaid to the investors.
During 2020, the Company issued a 3% interest promissory note of approximately $30,000 due on December 31, 2021 or earlier under certain events as defined in the promissory note in exchange for a vendor balance in accounts payable. The promissory note plus interest was settled during the year ended December 31, 2021.
On April 21, 2020, the Company received loan proceeds in the amount of approximately $22,000 under the Paycheck Protection Program (“PPP”), which was included in other current liabilities as of December 31, 2020. The PPP, established as part of the Coronavirus Aid, Relief and Economic Security Act (“CARES Act”), provided for loans to qualifying businesses for amounts up to 2.5 times of the average monthly payroll expenses of the qualifying business. Following the PPP guidelines, the Company filed for loan forgiveness in February 2021 and on March 4, 2021 the Small Business Administration approved the filing and forgave the loan. The Company recognized the gain on forgiveness in other income on the statements of operations during the year ended December 31, 2021.
NOTE 5 – STOCKHOLDERS’ EQUITY
COMMON STOCK
On November 10, 2021, the Company concluded its IPO of 2,750,000 units, (the “Units”), with each Unit consisting of one share of common stock, par value $0.0001 per share (the “Common Stock”) and one warrant (the “Warrants”) to purchase Common Stock at a combined public offering price of $6.00 per Unit. The Common Stock and the Warrants were immediately separable and issued separately but were purchased together in the IPO. The Warrants will have a per share exercise price of $6.00 and are exercisable immediately. The Warrants will expire five years from the date of issuance.
The Company received approximately $14,713,000 in net proceeds from the IPO after deducting the underwriting discount and commission and other IPO expenses payable by the Company of approximately $1,800,000.
On November 10, 2021, as a result of the completion of the IPO and as required under the terms of the 2015 Notes, the Company converted the entirety of the outstanding principal of $5,084,000 and interest accrued of $1,204,404 to 1,497,216 shares of common stock at the Conversion Price of $4.20 per share and issued the shares to the 2015 Note holders, fully satisfying the Company’s obligations.
Upon consummation of the above mentioned IPO, the company was required to issue 78,025 shares of Common Stock to a consulting firm for services provided that were related to the IPO. The Company calculated the value of the common stock using closing stock price on November 11, 2022, resulting in a fair value of approximately $365,000. Additionally, the Company was required to issue 72,727 warrants based on performance metrics achieved in 2021, the warrants have an exercise price of $5.50 with an expiration of five years from the date of issuance. The Company calculated the fair value of $1.25 each for these warrants using the Black-Scholes option pricing model on the date the consulting firm achieved the milestone, using the following assumptions: (a) fair value of $2.28 per share, (b) expected volatility of 90.81%, (c) dividend yield of 0%, (d) risk-free interest rate of 0.87%, and (e) expected life of 5 years, resulting in the fair value of approximately $91,000.
During the years ended December 31, 2021 and 2020 the Company issued 34,846 and 45,000 shares of common stock upon exercise of vested stock options.
WARRANTS
In connection with the short term notes issued in 2019, the Board of Directors approved the issuance of warrants. The Company issued 15,277 fully vested warrants as an incentive to investors with the rights to convert into a fixed number of shares of the Company’s common stock for an above market fixed price of $2.75 per share, exercisable, in whole or in part, for a period of 4 years from the date of issuance.
During 2019, milestone warrants were issued to certain executives of the Company totaling 407,272 units (“Penny Warrants”), these were valued on the date of grant at $0.0003 and will vest upon meeting certain milestones. The warrant may be exercised, in whole or in part upon the earliest to occur of: (i) following the Company’s initial public offering, the date on which the Company has a market capitalization of at least $50,000,000 for five consecutive business days; (ii) the closing of a Change of Control transaction with net proceeds to Company equity holders of at least $50,000,000; (iii) the date on which the Company receives a bona fide pre-money valuation from a third party investor of at least $50,000,000; (iv) the date on which the Holder’s continuous status as a Service Provider is terminated by the Company without Cause upon or within 12 months after a Change of Control; and (v) the date on which the Holder terminates his continuous status as a Service Provider for Good Reason within 12 months after a Change of Control.
Since these Penny Warrants have performance obligations to be met by the Company to become exercisable which are not met under any circumstance as of December 31, 2021, they are excluded from weighted-average shares outstanding in the net loss per share calculation.
In accordance with ASC Topic 480, Distinguishing Liabilities from Equity, as no derivative feature exists, the warrants issued to executives were classified as equity and the Company determined that as of December 31, 2021 and December 31, 2020 it is not likely that these warrants would vest and as such the value of the warrants would be deemed immaterial with no impact on the accompanying financial statements.
In connection with the IPO, the Company issued 2,750,000 Warrants, with a per share exercise price of $6.00 and exercisable immediately. The Warrants expire five years from the date of issuance.
Pursuant to the Underwriting Agreement dated November 10, 2021 between the Company and The Benchmark Company, LLC (the “Underwriter”) the Company granted the Underwriter a 30-day option to purchase up to an additional 412,500 shares of our Common Stock and/or Warrants to cover over-allotments. On consummation of the IPO, the Underwriter exercised the over-allotment option to purchase 412,500 Offering Warrants.
The Company also issued warrants to purchase Common Stock (7% of the number of Common Stock sold in IPO) to be issued to the Underwriter, as a portion of the underwriting compensation payable in connection with IPO. The Company issued 192,500 warrants, exercisable at a per share exercise price equal to $7.50 per share. The warrants will expire five years from the date of issuance and are subject to a 180-day lock-up period.
A summary of the outstanding warrants as of December 31, 2021 and 2020 is as follows:
| | | | | | | | | | | | | | | | | |
| Number of
shares | | Weighted
average exercise
price | | Weighted
average
remaining life (years) |
Outstanding and exercisable - December 31, 2020 and 2019 | 422,549 | | | $ | 0.11 | | | 2.12 years |
| Exercised | — | | | — | | | |
| Warrants issued | 3,355,000 | | | $ | 6.09 | | | |
Outstanding and exercisable – December 31, 2021 | 3,777,549 | | | $ | 5.42 | | | 4.45 years |
NOTE 6 – STOCK-BASED COMPENSATION
In 2015, the Company’s Board of Directors approved the HeartBeam, Inc. 2015 Equity Incentive Plan ("2015 Plan"), to attract and retain the best available personnel for positions of substantial responsibility, to provide additional incentive to employees, directors, and consultants, and to promote the success of the Company’s business. The 2015 Plan provides for the grant of stock options and restricted stock awards (“RSUs”) to purchase common stock. The Board of Directors approved 363,636 shares of common stock issuance under the 2015 Plan. On January 31, 2018, the Board of Directors added an additional 545,454 shares of common stock for issuance under the 2015 Plan. On June 15, 2021, the Board of Directors added an additional 727,272 shares of common stock for issuance under the 2015 Plan.
As of December 31, 2021, there were 234,439 shares available for issuance under the 2015 Plan.
STOCK OPTIONS
The following is a summary of stock option activity during the years ended December 31, 2021 and 2020:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of
options
outstanding | | Weighted
average
exercise
price (*) | | Average
remaining
contractual life
(in years) | | Aggregate
intrinsic value
(in thousands)
(**) |
| | | | | | | |
| Outstanding – December 31, 2019 | 297,560 | | | $ | — | | | 7.6 | | $ | 81 | |
| Options granted | 214,182 | | | 0.28 | | | | | |
| Options exercised | (45,000) | | | — | | | | | |
| | | | | | | |
Outstanding – December 31, 2020 | 466,742 | | | $ | 0.14 | | | 8.2 | | $ | 81 | |
Options granted | 679,495 | | | 3.22 | | | | | |
Forfeitures | (5,453) | | | 0.07 | | | | | |
Options exercised | (34,846) | | | — | | | | | |
| | | | | | | |
Outstanding – December 31, 2021 | 1,105,938 | | | 2.03 | | | 8.8 | | 1,535 | |
Exercisable – December 31, 2021 | 346,096 | | | $ | 0.57 | | | 7.3 | | $ | 878 | |
(*) $ - Indicates exercise price less than $0.01 per share
(**) Intrinsic value is based on the fair market value of the Company's common stock.
The Company estimates the fair values of stock options using the Black-Scholes option-pricing model on the date of grant. For the years ended December 31, 2021 and 2020, the assumptions used in the Black-Scholes option pricing model, which was used to estimate the grant date fair value per option, were as follows:
| | | | | | | | | | | |
| Year ended December 31, |
| 2021 | | 2020 |
| Weighted-average Black-Scholes option pricing model assumptions: | | | |
| Volatility | 90.01% - 106.22% | | 74.61% - 74.89% |
| Expected term (in years) | 5.69 - 5.93 | | 5.55 - 5.87 |
| Risk-free rate | 0.69% - 1.08% | | 0.25% - 0.39% |
| Expected dividend yield | $ | — | | | $ | — | |
| Weighted average grant date fair value per share | $2.07 - 3.44 | | $0.16 - 0.18 |
The following is a summary of stock-based compensation expense:
| | | | | | | | | | | | | | | |
| | | Year ended December 31, |
| | | | | 2021 | | 2020 |
| General and administration | | | | | $ | 164,933 | | | $ | 5,846 | |
| Research and development | | | | | 27,376 | | | 3,850 | |
| | | | | $ | 192,309 | | | $ | 9,696 | |
RESTRICTED STOCK UNITS
On December 14, 2021, the Company issued 30,000 shares of restricted stock awards to a consultant to provide services over the next two years. The total fair value of the issuances is $96,000. During the year ended December 31, 2021, the compensation expense for the RSUs was de minimis.
The following is a summary of non-vested RSUs award activity for the year ended December 31, 2021, we had no such activity in 2020: | | | | | | | | | | | | | | |
| | Number of Shares | | Weighted Average Grant Date Fair Value |
| Non-vested | | 30,000 | | | $ | 3.20 | |
NOTE 7 – RELATED PARTY TRANSACTIONS
During the course of business, the Company obtains accounting services from CTRLCFO, a firm in which the Company’s Chief Financial Officer has significant influence, as well as Hardesty, where he is a non-managing partner. The Company incurred accounting fees from these firms of approximately $88,000 and $82,000 during the years ended December 31, 2021 and 2020, respectively. As of December 31, 2021 and 2020, the Company had balances due to these firms amounting to approximately $1,000 and $15,000, respectively.
The Company’s Directors and Officers invested in the 2015 Notes of the Company, as did several consultants who provide services. On November 10, 2021, on completion of the IPO and as required under the terms of the 2015 Notes, the Company converted the entirety of the outstanding principal and interest accrued to the 2015 Notes to common stock, which included 586,256 shares issued to Directors and Officers, representing a principal amount of $1,927,000 and interest of $535,296 and 258,420 shares issued to consultants representing a principal amount of $923,000 and interest $162,363.
As of December 31, 2020, investments in the 2015 Notes from Directors and Officers was approximately $1,797,000, and investments from consultants was approximately $661,000.
NOTE 8 – COMMITMENTS AND CONTINGENCIES
On May 1, 2019, the Company entered into a month to month lease agreement for our headquarters. The agreement is for an undefined term and can be cancelled at any time, given one month’s notice by either party. The Company’s monthly rent expense associated with this agreement is approximately $1,440. The Company’s month to month headquarters lease is in the name of the Company’s Chief Executive Officer, and the cost is reimbursed monthly.
For the years ended December 31, 2021 and 2020, rent expense was approximately $17,000, for each year.
NOTE 9 - INCOME TAX
Income tax expense attributable to pretax loss from continuing operations differed from the amounts computed by applying the U.S. federal income tax rate of 21% to pretax loss from continuing operations as a result of the following:
| | | | | | | | | | | | | | | | | | | | |
| | For the Years ended December 31, |
| | 2021 | | 2020 |
| | | | | | |
| Computed “expected” tax benefit | | (930,000) | | 21.00 | % | | (224,100) | | 21.00 | % |
| | | | | | |
| Increase (reduction) in income taxes resulting from): | | | | | | |
| State tax, net of federal benefit | | (178,800) | | 4.04 | % | | (72,700) | | 6.77 | % |
| Permanent items | | 393,400 | | (8.88) | % | | 1,700 | | (0.16) | % |
| State research and development credits | | (6,200) | | 0.14 | % | | (2,500) | | 0.23 | % |
| Change in valuation allowance | | 726,800 | | (16.41) | % | | 297,600 | | (27.84) | % |
| Other | | (5,200) | | 0.11 | % | | — | | — | % |
| | | | | | |
| Total | | — | | — | % | | — | | — | % |
The tax effects of temporary differences that give rise to significant portions of the deferred tax assets and liabilities are presented below as of December 31:
| | | | | | | | | | | | | | |
| | For the Years ended December 31, |
| | 2021 | | 2020 |
Deferred tax assets (liabilities): | | | | |
Net operating loss carryforwards | | $ | 1,982,000 | | | $ | 1,317,000 | |
Research and development credits | | 33,200 | | | 27,000 | |
Other | | 59,500 | | | 3,900 | |
Total deferred tax assets | | 2,074,700 | | | 1,347,900 | |
| | | | |
Valuation Allowance | | (2,074,700) | | | (1,347,900) | |
| | | | |
Net Deferred Tax Assets | | — | | | — | |
Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain. Accordingly, the net deferred tax assets have been fully offset by a valuation allowance. The valuation allowance increased by approximately $726,800 for the period ended December 31, 2021.
As of December 31, 2021, the Company had net operating loss carryforwards for federal and state income tax purposes; each are approximately $7,100,000. If not utilized, these net federal and state operating loss carryforwards will expire beginning in 2035. The 20-year limitation was eliminated for losses generated after January 1, 2018, giving the taxpayer the ability to carry forward losses indefinitely. However, net operating losses will now be limited to 80 percent of taxable income. In assessing the realizability of the deferred tax assets, management considers whether it is more likely than not that some portion or all of the deferred tax assets will not be realized. Management considers the scheduled reversal of deferred tax liabilities, projected future taxable income, net operating loss carryback potential and tax planning strategies in making these assessments.
As of December 31, 2021, the Company has federal and state tax credit carryforwards of $7,882 and $50,042, respectively. The state tax credit carryforwards do not expire.
Under Section 382 of the Internal Revenue Code of 1986, as amended, the Company's ability to utilize NOLs or other tax attributes such as research tax credits, in any taxable year may be limited if the Company experiences, or has experienced, an "ownership change." A Section 382 "ownership change" generally occurs if one or more stockholders or groups of stockholders, who own at least 5% of the Company's stock, increase their ownership by more than 50 percentage points
over their lowest ownership percentage within a rolling three-year period. Similar rules may apply under state tax laws. The Company may in the future experience, one or more Section 382 "ownership changes." If so, the Company may not be able to utilize a material portion of its NOLs and tax credits, even if the Company achieves profitability.
The Company’s policy to recognize interest and penalties associated with unrecognized tax benefits as part of the income tax provision and include accrued interest and penalties with the related income tax liability on the Company’s balance sheet. To date, the Company has not recognized any interest and penalties in its statements of operations, nor has it accrued for or made payments for interest and penalties associated with unrecognized tax benefits.
The Company files federal and state income tax returns with varying statutes of limitations. The tax years from inception through 2021 remain open to examination due to the carryover of unused net operating losses and tax credits.
NOTE 10 - SUBSEQUENT EVENTS
On January 31, 2022, the Company entered into a partnership agreement (the “Partnership Agreement”) with LIVMOR, Inc. (“LIVMOR”), a digital health solutions company, to build a Company-branded version of the LIVMOR’s Halo+ FDA cleared turnkey solution for RPM to connect physicians and patients. The Company-branded version (“HeartBeam Platform”) of LIVMOR’s cloud-based remote monitoring portal will be an FDA registered Class 1 system and fully compliant with FDA standards for cybersecurity, software engineering and human factors and includes the prerequisite infrastructure for industry-leading solutions for telehealth. The Partnership Agreement also outlines rights and responsibilities for the customization of the HeartBeam Platform by LIVMOR with senior executives of both companies supervising the project. The Company’s Partnership Agreement with LIVMOR further support’s the Company’s project schedule for the FDA submission of its first product, an easy-to-use heart attack detection software solution, in an Emergency Department setting. In addition, the Partnership Agreement provides a statement of work for development of the Company’s product, project management supervised by senior executives of the Company and LIVMOR, and regulatory support assistance for HeartBeam Platform by LIVMOR to obtain FDA clearance. Per the Partnership Agreement, the Company and LIVMOR have the right to enter into additional agreements as needed in order to further the Company’s development of its products.
On February 18, 2022, the Company entered into a stock purchase agreement pursuant to which the Company agreed to issue and sell (“Private Placement”) to OpenSky Opportunities Fund Ltd. (“Purchaser”) an aggregate of 58,000 units (“Units”), with each Unit consisting of one share of common stock, par value $0.0001 per share (“Common Stock”) and one warrant (the “Warrants”) to purchase one share of Common Stock at a combined price of $6 per Unit. The Common Stock and the Warrants were immediately separable and issued separately but were purchased together in the Private Placement. The Units, the Common Stock and the Warrants issued pursuant to the stock purchase agreement shall be referred to as the “Securities”. We received $348,000 in proceeds from the Private Placement. The Warrants issued with the Private Placement will have a per share exercise price of $6.00 and are exercisable immediately subject to a 180-Day lock up. The Warrants will expire five years from the date of issuance. The Stock Purchase Agreement contains customary representations and warranties of the parties.
On March 7, 2022 the Company entered into a professional services agreement (the “Triple Ring Agreement”) with Triple Ring Technologies, Inc. (“Triple Ring”), a co-development company, to assist in the design and development of our telehealth complete solution 3D vector ECG collection device for remote heart attack or MI monitoring. The Triple Ring Agreement is a five-phase expedited device development project scheduled to be completed in the fourth quarter of 2022 for a 510k submission to the FDA.
Under the terms of the Triple Ring Agreement, the joint project will include our telehealth 3D vector ECG collection device builds for design verification and validation, device packaging, and a manufacturing technology transfer to a contract manufacturer to be named later.
The agreements with LIVMOR and Triple Ring include commitments in 2022 of approximately $3.0 million, of which approximately $1.4 million have been made to date. In addition, there will be license fees on the future commercial use of the platform.